

In this issue of Blood, demonstrate the role of complement activation in antiphospholipid syndrome (APS) and a high prevalence of germline mutations in complement regulatory genes in patients with catastrophic APS (CAPS).1
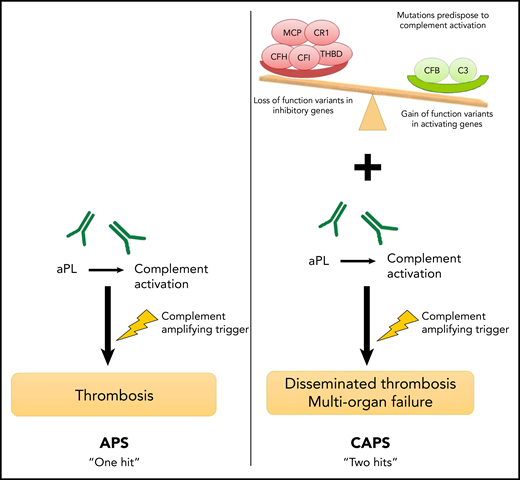
A proposed framework for differences in the mechanisms of APS and CAPS. In APS, a complement-amplifying trigger induces thrombosis in the presence of aPL antibodies such as the lupus anticoagulant, anticardiolipin, or β2GP-I antibodies. The presence of an underlying germline mutation in complement regulatory genes may increase the risk of developing CAPS, a more severe subtype.
A proposed framework for differences in the mechanisms of APS and CAPS. In APS, a complement-amplifying trigger induces thrombosis in the presence of aPL antibodies such as the lupus anticoagulant, anticardiolipin, or β2GP-I antibodies. The presence of an underlying germline mutation in complement regulatory genes may increase the risk of developing CAPS, a more severe subtype.
APS is an acquired thrombophilic state in which thrombosis or obstetric morbidity are induced by antiphospholipid (aPL) antibodies such as the lupus anticoagulant, anticardiolipin, or β2-glycoprotein-I (β2GP-I) antibodies.2,3 Patients may be positive for 1, 2, or all 3 antibodies, with the so-called triple-positive syndrome conferring the highest risk of recurrent thrombosis. The transient presence of these antibodies without evidence of APS is common in the general population in which up to 10% of the normal blood donor pool exhibits one or more of these antibodies on routine testing.4 Conversely, a subset of patients with APS (those with CAPS) will experience thrombotic events and multisystem organ failure. The working criteria for CAPS include a history of APS or aPL antibodies with 3 or more new organ thromboses within 1 week, histologic confirmation of microthrombus, and exclusion of other causes.5 Therapy for APS is long-term anticoagulation, but recurrent thromboses are an ever worrisome issue. Although complement activation has previously been implicated in the pathogenesis of APS and CAPS, the use of complement inhibitor therapy to treat this patient population has been limited to case reports.6-8
In the Chaturvedi et al study, the authors evaluate complement activity in patients with APS, CAPS, and systemic lupus erythematosus (SLE) by using the modified Ham (mHam) test. This assay uses a line of TF-1 cells that are engineered to lack PIGA and consequently lack membrane proteins that protect cells from activated complement. These PIGA null cells bind terminal complement pathway proteins and are killed in the presence of activated complement.9 The authors found that although 35.6% of patients with thrombotic APS had a positive mHam test, more than 85% of the patients with CAPS had a positive mHam test, which indicates increased complement activity. Furthermore, patients who were triple positive for aPL antibodies or those with recurrent thrombosis were more likely to have a positive mHam test compared with controls.
The authors confirm by flow cytometry that sera from APS patients induces terminal complement C5b-9 deposition on the PIGA null TF-1 cells, which could be blocked by using complement inhibitor therapy. Interestingly, inhibition of complement factor D, which mediates the alternative complement pathway, was unable to prevent cell killing in the mHam test or C5b-9 deposition, further localizing aPL antibody activation to the classical complement pathway.
In what may be the most striking finding of the study, the authors sequenced 15 known genes related to complement function and found that although APS patients had a mutation rate comparable to that of patients with SLE and normal controls, 60% of those with CAPS had an identifiable pathogenic mutation, a rate as high as or higher than that seen in atypical hemolytic uremic syndrome.
The authors provide a framework for their findings (see figure), which suggests that although APS requires a single complement-amplifying trigger, CAPS is more likely to occur in a patient who experiences the same trigger in the context of heritable mutations that increase the risk of complement activation. These mutations can result in proteins with either a loss of function of complement-inhibitory proteins or a gain of function in complement-activating proteins.
These findings raise several questions and suggest new areas for investigation. Should APS patients undergo mutational analysis to assess risk for CAPS? Given the significant morbidity and mortality associated with CAPS, should patients receive ongoing complement inhibitor therapy in addition to anticoagulation if the mHam test suggests increased complement activity or if flow cytometry shows increased terminal complement C5b-9 deposition?
The need for long-term anticoagulation to reduce recurrent thrombosis is clear, but the question of which anticoagulant is ideal has been intensely studied in recent years. The direct oral anticoagulants (DOACs) have significantly improved our anticoagulation management strategies in most thrombotic conditions, but their use in APS was put into question when the TRAPS trial was terminated prematurely because of an increase in both thromboembolic events and major bleeding in the rivaroxaban arm when compared with the warfarin arm.10 Given that initial anticoagulation choices are often made without knowledge of aPL antibody test results, it is unclear whether a patient receiving DOAC therapy who is subsequently found to have a positive aPL antibody test should be switched to warfarin when they seem to be clinically stable on their current therapy. Could the mHam test be used to determine which patients must be treated with warfarin and those patients who may be safely treated with a DOAC?
It will be interesting to see whether the mHam test and mutational testing will identify a subset of patients who have a low risk for recurrence and may be able to stop long-term anticoagulation.
The next step will be to evaluate these findings in larger cohorts of patients along with additional clinical studies to determine the efficacy of complement inhibitor therapy in patients with APS or CAPS. It may take years to answer these questions, but the authors have significantly advanced our understanding of this rare and challenging condition and opened new doors for potential investigation.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal