

Key Points
We elucidate a previously unknown role of GPS2 in erythroid differentiation by using human CD34+ cells and GPS2-knockout mice.
GPS2 interacts with EKLF and prevents its degradation, providing a new understanding of KLF1 insufficiency–caused hematologic disorders.

Abstract
Erythropoiesis is a complex multistage process that involves differentiation of early erythroid progenitors to enucleated mature red blood cells, in which lineage-specific transcription factors play essential roles. Erythroid Krüppel-like factor (EKLF/KLF1) is a pleiotropic erythroid transcription factor that is required for the proper maturation of the erythroid cells, whose expression and activation are tightly controlled in a temporal and differentiation stage-specific manner. Here, we uncover a novel role of G-protein pathway suppressor 2 (GPS2), a subunit of the nuclear receptor corepressor/silencing mediator of retinoic acid and thyroid hormone receptor corepressor complex, in erythrocyte differentiation. Our study demonstrates that knockdown of GPS2 significantly suppresses erythroid differentiation of human CD34+ cells cultured in vitro and xenotransplanted in nonobese diabetic/severe combined immunodeficiency/interleukin-2 receptor γ-chain null mice. Moreover, global deletion of GPS2 in mice causes impaired erythropoiesis in the fetal liver and leads to severe anemia. Flow cytometric analysis and Wright-Giemsa staining show a defective differentiation at late stages of erythropoiesis in Gps2−/− embryos. Mechanistically, GPS2 interacts with EKLF and prevents proteasome-mediated degradation of EKLF, thereby increasing EKLF stability and transcriptional activity. Moreover, we identify the amino acids 191-230 region in EKLF protein, responsible for GPS2 binding, that is highly conserved in mammals and essential for EKLF protein stability. Collectively, our study uncovers a previously unknown role of GPS2 as a posttranslational regulator that enhances the stability of EKLF protein and thereby promotes erythroid differentiation.
Introduction
Erythroid Krüppel-like factor (EKLF, encoded by the KLF1 gene) is a well-studied erythroid-specific transcription factor with preferential activation of genes for components of the red blood cell membrane and cytoskeleton, heme and globin synthesis, and cell-cycle regulation.1 Mice lacking EKLF develop fatal anemia during fetal liver erythropoiesis, due to a defect in the maturation of red blood cells, and die around embryonic day 14 (E14).2 In addition, loss-of-function mutations in the human KLF1 gene have been associated with various kinds of human hematologic disorders.3 The evidence that KLF1 haploinsufficiency perturbs the regulation of erythropoiesis underlines the requirement for tight regulation of EKLF levels.3 EKLF expression and activation are regulated by mechanisms such as control of EKLF RNA transcription, protein stability, subcellular localization, and posttranslational modifications.4 It has been previously reported that the stability of EKLF is regulated by the ubiquitin-proteasome pathway.5 EKLF is ubiquitinated in vivo; however, its modification does not rely on a particular internal lysine.5 The transactivation domain 1 (TAD1) from EKLF has been shown to be able to form noncovalent interactions with ubiquitin and contribute to ubiquitin-mediated degradation of EKLF.6 In addition, the 2 conserved PEST sequences in EKLF are not required for ubiquitination, but they may serve as docking sites for proteasome-mediated unfolding of the EKLF protein prior to degradation.5 Moreover, Ppm1b, a serine-threonine protein phosphatase, is reported to interact with EKLF and play a positive role in erythroid differentiation by increasing the stability of EKLF protein.7 Thus, the posttranslational control mechanism for regulation of EKLF protein level may be critical to the role of EKLF in red blood cell development. However, the regulating proteins and the underlying mechanisms are still little known.
G-protein pathway suppressor 2 (GPS2) was originally identified while screening for suppressors of lethal G-protein subunit-activating mutations in the yeast pheromone response pathway.8 GPS2 is mainly studied as a transcriptional cofactor, which can act as both a transcriptional activator and a repressor. Multiple functional interactions have been reported between GPS2 and transcriptional regulators, including the nuclear receptor corepressor 1 (NCOR1) and silencing mediator of retinoic acid and thyroid hormone receptor corepressor complex,9 the histone acetyltransferase p300,10 and numerous DNA-binding transcription factors.11-18 Furthermore, a critical nontranscriptional role of GPS2 in the cytoplasm has been identified.15 GPS2 may directly inhibit Ubc13 enzymatic activity and modulate the Ubc13-mediated K63 ubiquitination events, which is functionally critical for many pathways, including tumor necrosis factor receptors, Toll-like receptors, B-cell antigen receptors, and phosphatidylinositol 3-kinase/AKT signaling pathways.15,19,20 The use of whole-body and tissue-specific GPS2-knockout mice has provided evidences for essential functions of GPS2 in embryonic development,21 inflammation,22 metabolism,18,19,22 and B-cell development.20 GPS2 is highly expressed in basophilic erythroblasts and upregulated in reticulocytes purified from fetal mice according to a study of the ontogeny of erythroid gene expression.23 Moreover, as a component of the NCOR1-HDAC3 complex, GPS2 may contribute to the role of NCOR1 in erythroid differentiation.24-27 We therefore speculate that GPS2 might be involved in the regulation of erythroid differentiation.
In this paper, we investigate the contribution of GPS2 to erythroid differentiation using GPS2-silenced human umbilical cord blood (UCB) CD34+ cells and GPS2-knockout mice. Our data indicate that GPS2 is critical for erythroid differentiation, especially erythroblast maturation. Mechanistic studies reveal that GPS2 interacts with EKLF and prevents proteasome-mediated degradation of EKLF protein, thereby increasing the availability of EKLF to activate transcription.
Materials and methods
Mice
Gps2+/− mice were generated using zinc-finger nuclease (ZFN) technology on a C57/BL/6N background at Institute of Laboratory Animals Science, Chinese Academy of Medical Sciences and Peking Union Medical College. Custom-made ZFN plasmids designed for Gps2 gene were obtained from Sigma-Aldrich. The ZFN binding and cutting sites are 5′- CAGCATCATGCCCGCactcctGGAGCGCCCCAAGCT-3′. 8-week-old 129S2/Sv mice were purchased from the Beijing Vital River Laboratory Animal Technology. Gps2+/− C57BL/6N mice were crossed with wild-type (WT) 129S2/Sv mice to obtain the Gps2+/− mice with a mixed genetic background (50% C57BL/6N: 50% 129S2/Sv). All mouse strains were maintained in specific pathogen–free conditions at the animal facility of our institute. All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of our institute.
Cell culture
Flow cytometry and cell sorting
A detailed procedure is described in supplemental Data.
Methylcellulose colony-forming assays
M3434 and H4435 methylcellulose medium from StemCell Technologies were used according to the manufacturer’s instructions.
Benzidine staining assay
Benzidine staining assay was performed as described.30 A detailed protocol is provided in supplemental Materials.
Mouse xenotransplantation
Freshly isolated human UCB CD34+ cells were cultured in StemSpan SFEM II Medium supplemented with recombinant human stem cell factor (100 ng/mL), recombinant human Flt-3 ligand (100 ng/mL), and recombinant human thrombopoietin (50 ng/mL) for 24 hours and then infected with lentivirus (multiplicity of infection = 20, 2 infections 12 hours apart). At the end of the second infection, 1 × 106 of transduced cells were IV injected into sublethally irradiated (250 cGy, 12 hours before injection) 8-week-old nonobese diabetic/severe combined immunodeficiency/interleukin-2 receptor γ-chain null (NSG; Biocytogen, B-CM-002) mice. Recipients were sacrificed at either 3 or 20 weeks after transplantation, and engraftment in bone marrow (BM) was analyzed.
Luciferase reporter assay
Cells were lysed in passive lysis buffer, and luminescence levels were measured using the Dual-Luciferase Reporter Assay System (Promega, E1910) according to the manufacturer’s instructions.
CHX chase assay
Cells were incubated in 200 μg/mL cycloheximide (CHX; Cell Signaling Technology, 2112) for indicated times before being lysed in 2 × Laemmli sample buffer. Protein level was determined by western blot analysis.
Western blot, immunoprecipitation, and ubiquitination assays
Detailed procedures are described in supplemental Materials.
Statistical analyses
Statistical analyses were performed with GraphPad Prism software version 7. Data are shown as mean ± standard error of the mean (SEM). A 2-tailed unpaired Student t test or Mann-Whitney test was used to compare the mean of 2 groups, and a 2-way analysis of variance (ANOVA) was used for multiple comparisons. P < .05 was considered statistically significant.
Results
GPS2 expression is upregulated during erythroid differentiation of CD34+ cells
To address the role of GPS2 in erythroid differentiation, we first investigated the expression of GPS2 in UCB hematopoietic stem/progenitor cells (HSPCs). Hematopoietic stem cells (HSCs), common myeloid progenitors (CMPs), granulocyte/macrophage progenitors (GMPs), and megakaryocyte/erythroid progenitors (MEPs) were isolated from UCB as previously described (Figure 1A).31 Real-time polymerase chain reaction (PCR) analysis showed that CMPs and MEPs had a similar level of GPS2 transcript compared with HSCs, whereas GMPs possessed an approximately twofold lower level of GPS2 transcript (Figure 1B). The expression of GPS2 in MEPs was almost comparable with GATA1 (Figure 1B). In addition, GPS2 expression was much higher in burst-forming unit-erythroids (BFU-Es) than in colony-forming unit granulocytes/macrophages (CFU-GMs) cultured from UCB CD34+ cells (Figure 1C-D). We next examined the expression of GPS2 during erythroid differentiation of UCB CD34+ cells (Figure 1E; supplemental Figure 1). The results showed that GPS2 expression level was significantly upregulated during erythroid differentiation as measured by real-time PCR and western blot analysis (Figure 1F-G).

GPS2 expression is upregulated during erythroid differentiation of CD34+cells. (A) Flow cytometry gating strategy for HSPCs in CD34+ cells isolated from human UCB: HSCs (CD34+CD38−), CMPs (CD34+CD38+CD123+CD45RA−), GMPs (CD34+CD38+CD123+CD45RA+), and MEPs (CD34+CD38−CD123−CD45RA−). (B) The relative expression of GPS2 and GATA1 in HSCs, CMPs, GMPs, and MEPs isolated from human UCB was analyzed by real-time PCR (normalized to GAPDH levels). (C) Representative microscopy images of BFU-Es and CFU-GMs cultured from human UCB CD34+ cells. (D) Real-time PCR analysis of GPS2 and GATA1 expression in BFU-Es and CFU-GMs (normalized to GAPDH levels). (E-G) Human UCB CD34+ cells were isolated and cultured for 2 days in expansion medium, which was then changed to erythroid differentiation medium for the indicated times. Flow cytometry plots showing the expression of CD71 and glycophorin A (GPA) (E). Real-time PCR (F) and western blot (G) analysis of the expression of GPS2. All values are mean ± SEM (n = 3 replicates). *P < .05, ***P < .001; 2-tailed unpaired t test.
GPS2 expression is upregulated during erythroid differentiation of CD34+cells. (A) Flow cytometry gating strategy for HSPCs in CD34+ cells isolated from human UCB: HSCs (CD34+CD38−), CMPs (CD34+CD38+CD123+CD45RA−), GMPs (CD34+CD38+CD123+CD45RA+), and MEPs (CD34+CD38−CD123−CD45RA−). (B) The relative expression of GPS2 and GATA1 in HSCs, CMPs, GMPs, and MEPs isolated from human UCB was analyzed by real-time PCR (normalized to GAPDH levels). (C) Representative microscopy images of BFU-Es and CFU-GMs cultured from human UCB CD34+ cells. (D) Real-time PCR analysis of GPS2 and GATA1 expression in BFU-Es and CFU-GMs (normalized to GAPDH levels). (E-G) Human UCB CD34+ cells were isolated and cultured for 2 days in expansion medium, which was then changed to erythroid differentiation medium for the indicated times. Flow cytometry plots showing the expression of CD71 and glycophorin A (GPA) (E). Real-time PCR (F) and western blot (G) analysis of the expression of GPS2. All values are mean ± SEM (n = 3 replicates). *P < .05, ***P < .001; 2-tailed unpaired t test.
Knockdown of GPS2 blocks human erythroid differentiation
To explore the roles GPS2 during human erythroid differentiation, we used a lentiviral short hairpin RNA (shRNA)–mediated knockdown approach in human UCB CD34+ cells (Figure 2A). An >80% knockdown efficiency was achieved with either of the 2 GPS2 shRNA compared with a scrambled shRNA at day 5 of erythropoietin (EPO)–induced differentiation (Figure 2B-C). We next examined the effects of GPS2 knockdown on EPO-induced erythroid differentiation. The results showed that CD34+ cells transduced with GPS2 shRNA lentivirus generated a significantly reduced proportion of CD71+GPA+ cells at days 3 and 5 of differentiation (Figure 2D; supplemental Figure 2A). Consistently, cytospin analyses showed that most of the GPS2-knockdown cells are still at the early stages of differentiation while control cells progressively differentiated to orthochromatic erythroblasts (supplemental Figure 2B). Knockdown of GPS2 inhibited the proliferation of CD34+ cells cultured in the erythroid differentiation medium, blocking the cells in G0/G1 phase (supplemental Figure 3A-B). In addition, downregulation of GPS2 decreased the percentage of benzidine-positive cells (Figure 2E-F). Consistently, knockdown of GPS2 significantly reduced the messenger RNA (mRNA) levels of erythroid genes, including ALAS2, GPA, BCL11A, NFE2, HBA1, and HBB, but not GATA1 and EKLF, in fluorescence-activated cell sorter (FACS)–sorted CD71+GPA+ cells at day 3 of differentiation (Figure 2G). Furthermore, CFU assays showed that knockdown of GPS2 resulted in a remarkable decrease in the number and proportion of BFU-Es (Figure 2H-I). In contrast, the number of CFU-GMs and CFU-granulocytes, erythrocytes, macrophages, megakaryocytes (CFU-GEMMs) was not altered (Figure 2H). We also investigated whether knockdown of GPS2 affects the generation of hematopoietic progenitor cells (MEPs in particular) and found that GPS2-silenced CD34+ cells cultured in the expansion medium had a similar proportion of CMPs, GMPs, and MEPs compared with control cells (supplemental Figure 4A-C). Taken together, these results suggest that GPS2 is essential for erythroid differentiation of human UCB CD34+ cells.
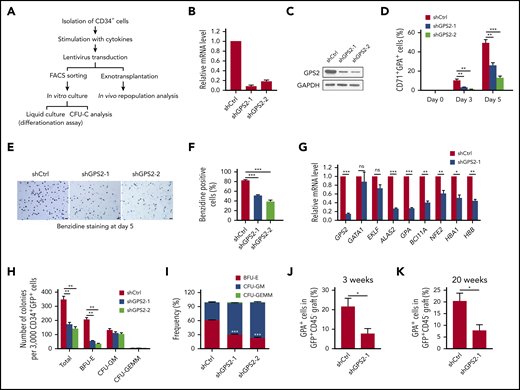
Knockdown of GPS2 blocks human erythroid differentiation. (A) Assays used to test the effect of GPS2 protein on erythropoiesis. CFU-C, CFU in culture. (B-G) CD34+GFP+ cells were selected from human UCB CD34+ cells infected with GPS2 or control (Ctrl) shRNA lentivirus and cultured in the erythroid differentiation medium for the indicated times. Knockdown efficiency of GPS2 was detected by real-time PCR (B) and western blot analysis (C) at day 5. The proportion of CD71+GPA+ cells was analyzed by flow cytometry at days 0, 3, and 5 (D). Hemoglobin production was analyzed by benzidine staining at day 5. Representative images of benzidine-stained cells were shown; scale bars, 20 μm (E). The percentage of benzidine-positive cells was calculated (F). mRNA levels of the indicated erythroid genes in FACS-sorted CD71+GPA+ cells were analyzed by real-time PCR at day 3 (G). (H-I) Clonogenic capacity of CD34+ cells transduced with GPS2 or control shRNA lentivirus. The number (H) and frequency (I) of colonies are represented. All values are mean ± SEM (n = 3 replicates). (J-K) Human CD34+ cells transduced with GPS2 or control shRNA lentivirus were IV injected into sublethally irradiated NSG mice. The percentage of GPA+ cells within the GFP+CD45− population in BM was determined at 3 (J) and 20 (K) weeks after transplantation (3 weeks, n = 6/group; 20 weeks, n = 4/group, mean ± SEM). *P < .05, **P < .01, ***P < .001; 2-tailed unpaired t test.
Knockdown of GPS2 blocks human erythroid differentiation. (A) Assays used to test the effect of GPS2 protein on erythropoiesis. CFU-C, CFU in culture. (B-G) CD34+GFP+ cells were selected from human UCB CD34+ cells infected with GPS2 or control (Ctrl) shRNA lentivirus and cultured in the erythroid differentiation medium for the indicated times. Knockdown efficiency of GPS2 was detected by real-time PCR (B) and western blot analysis (C) at day 5. The proportion of CD71+GPA+ cells was analyzed by flow cytometry at days 0, 3, and 5 (D). Hemoglobin production was analyzed by benzidine staining at day 5. Representative images of benzidine-stained cells were shown; scale bars, 20 μm (E). The percentage of benzidine-positive cells was calculated (F). mRNA levels of the indicated erythroid genes in FACS-sorted CD71+GPA+ cells were analyzed by real-time PCR at day 3 (G). (H-I) Clonogenic capacity of CD34+ cells transduced with GPS2 or control shRNA lentivirus. The number (H) and frequency (I) of colonies are represented. All values are mean ± SEM (n = 3 replicates). (J-K) Human CD34+ cells transduced with GPS2 or control shRNA lentivirus were IV injected into sublethally irradiated NSG mice. The percentage of GPA+ cells within the GFP+CD45− population in BM was determined at 3 (J) and 20 (K) weeks after transplantation (3 weeks, n = 6/group; 20 weeks, n = 4/group, mean ± SEM). *P < .05, **P < .01, ***P < .001; 2-tailed unpaired t test.
Knockdown of GPS2 suppresses erythroid cell generation in vivo in NSG mice
To examine whether GPS2 regulates human erythroid differentiation in vivo, human UCB CD34+ cells infected with GPS2-knockdown or control lentivirus were transplanted into sublethally irradiated NSG mice. Aliquots of cells used for transplantation were also cultured for 72 hours and assessed for GFP expression. Control and GPS2 shRNA–transduced cells showed a similar percentage of GFP expression (supplemental Figure 5A). However, knockdown of GPS2 led to a trend toward lower engraftment of CD45+GFP+ cells compared with control (supplemental Figure 5B), indicating that GPS2-knockdown cells might have a disadvantage during in vivo repopulation. We next carried out flow cytometric analysis of the proportion of human erythroid cells (CD45−GPA+) and found that the percentage of GFP+ erythroid cells at weeks 3 and 20 were both significantly reduced in the BM from mice receiving GPS2 shRNA–transduced cells (Figure 2J-K; supplemental Figure 5C). These findings indicate that GPS2 is required for the effective generation of erythroid cells in vivo. In addition, mice receiving GPS2 shRNA–transduced cells displayed a lower proportion of B cells (CD19+) but a higher proportion of myeloid cells (CD33+) in the BM (supplemental Figure 5D), which is consistent with previous reports that GPS2 is involved in the regulation of B-cell development20 and may account for the lower engraftment caused by GPS2 knockdown.
Definitive erythropoiesis is impaired in the GPS2-deficient fetal liver
To further study the functions of GPS2 in vivo, we generated whole-body Gps2-deficient (Gps2−/−) mice on the C57BL/6N background (supplemental Figure 6A-C). Similar to a previous report,21 we found that deficiency of GPS2 induced in utero lethality in 100% of embryos within the embryonic day 10.5 (E10.5) and E13.5 developmental stages (supplemental Figure 6D). Less than 10% viable homozygous embryos were observed at E12.5 or later, and these embryos were growth retarded and showed a smaller overall size compared with their littermates (supplemental Figure 6E). The paleness of the surviving Gps2−/− embryos suggests anemia (supplemental Figure 6E).
The early lethality of the Gps2−/− embryos makes the total contribution of GPS2 in hematopoiesis difficult to study in vivo. We thus generated compound heterozygous mice by crossing Gps2+/− mice and Gps2+/+ mice in the C57BL/6N and 129S2/Sv backgrounds, respectively. Embryonic death of Gps2−/− mice on the C57BL/6N:129S2/Sv background was delayed to E13.5 (Figure 3A), and severe anemia was observed in approximately all E12.5 embryos (Figure 3B). Gps2−/− fetal liver was noticeably smaller in size at E12.5 with reduced cell number (Figure 3B-C). The number of Ter119+ cells in Gps2−/− fetal livers was significantly reduced, but the number of nonerythroid hematopoietic cells (CD45+) was only mildly affected (Figure 3D-E). Analysis of E12.5 fetal liver cytospin preparations by Wright-Giemsa staining showed a disproportionate abundance of immature erythroblasts, such as proerythroblasts, in Gps2−/− fetal livers compared with WT controls (Figure 3F). These results confirm that definitive erythropoiesis is impaired in the GPS2-deficient fetal liver. In addition, cytospin analysis of peripheral blood from E12.5 embryos showed a significantly decreased proportion of enucleated erythrocytes in Gps2−/− embryos compared with littermate controls, which suggests a possible defect in primitive erythropoiesis (Figure 3G-H).
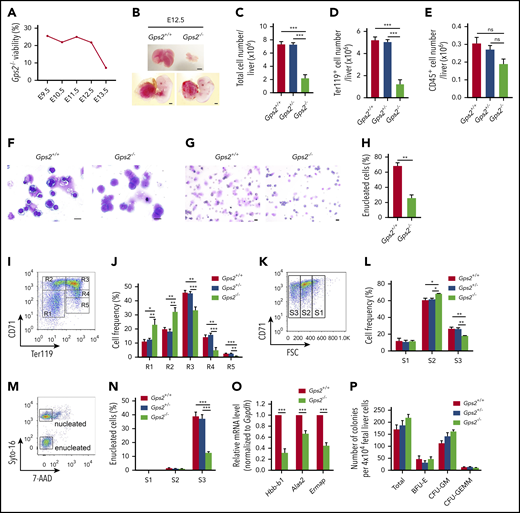
Definitive erythropoiesis is impaired in the GPS2-deficient fetal liver. (A) Viability of Gps2−/− embryos on the C57BL/6N:129S2/Sv background. Embryos from Gps2+/− intercrosses were harvested at stages E9.5 to E13.5. The percentages reflect the numbers of live Gps2−/− embryos with respect to all embryos harvested in litters at each gestational stage. (B) Representative pictures of E12.5 Gps2+/+ and Gps2−/− embryos (bottom) and fetal livers (top). Scale bars, 1 mm. (C-E) Total cell numbers (C), Ter119+ erythroid cell numbers (D), and CD45+ hematopoietic cell numbers (E) in fetal livers from Gps2+/+ (n = 17), Gps2+/− (n = 21), and Gps2−/− (n = 7) embryos at E12.5. Data are presented as mean ± SEM; Mann-Whitney test. (F) Wright-Giemsa staining of E12.5 Gps2+/+ and Gps2−/− fetal liver cytospin preparations. Scale bars, 10 μm. (G) Wright-Giemsa staining of peripheral blood cytospins from Gps2+/+ and Gps2−/− embryos at E12.5. Scale bars, 20 μm. (H) The percentage of enucleated red blood cells in peripheral blood cytospins from Gps2+/+ and Gps2−/− embryos at E12.5 (n = 3/group, mean ± SEM). Two-tailed unpaired t test. (I) Representative flow cytometry profiles of R1 to R5 erythroblast populations labeled with CD71 and Ter119 in fetal liver from Gps2+/+ embryos at E12.5. (J) The frequency of R1 to R5 cells in Gps2+/+ (n = 5), Gps2+/− (n = 8), and Gps2−/− (n = 4) E12.5 fetal livers. Data are presented as mean ± SEM; 2-tailed unpaired t test. (K) Representative flow cytograms of Gps2+/+ Ter119hi fetal liver cells (E12.5) separated into 3 populations (S1, S2, and S3) based on the forward light scatter (FSC) profile. (L) The frequency of S1 to S3 cells in Ter119hi fetal liver cells from each embryo at E12.5 (Gps2+/+, n = 5; Gps2+/−, n = 8; Gps2−/−, n = 4). Data are presented as mean ± SEM; 2-tailed unpaired t test. (M) Representative flow cytometry of enucleated cells in the S1 to S3 populations using Cyto-16 for nuclei and 7-aminoactinomycin D (7-AAD) for cell viability. (N) Percentages of enucleated cells in S1, S2, and S3 populations (Gps2+/+, n = 6; Gps2+/−, n = 11; Gps2−/−, n = 6). Data are presented as mean ± SEM; 2-tailed unpaired t test. (O) Real-time PCR analysis of the indicated erythroid genes in FACS-sorted CD71+Ter119+ cells from Gps2+/+ and Gps2−/− E12.5 fetal liver. Values are mean ± SEM (n = 3 replicates); 2-tailed unpaired t test. (P) Quantification of BFU-E, CFU-GM, and CFU-GEMM colonies from Methocult cultures of 4 × 104Gps2+/+, Gps2+/−, and Gps2−/− E12.5 fetal liver cells (n = 4/group). Values are mean ± SEM. *P < .05, **P < .01, ***P < .001; ns, not significant.
Definitive erythropoiesis is impaired in the GPS2-deficient fetal liver. (A) Viability of Gps2−/− embryos on the C57BL/6N:129S2/Sv background. Embryos from Gps2+/− intercrosses were harvested at stages E9.5 to E13.5. The percentages reflect the numbers of live Gps2−/− embryos with respect to all embryos harvested in litters at each gestational stage. (B) Representative pictures of E12.5 Gps2+/+ and Gps2−/− embryos (bottom) and fetal livers (top). Scale bars, 1 mm. (C-E) Total cell numbers (C), Ter119+ erythroid cell numbers (D), and CD45+ hematopoietic cell numbers (E) in fetal livers from Gps2+/+ (n = 17), Gps2+/− (n = 21), and Gps2−/− (n = 7) embryos at E12.5. Data are presented as mean ± SEM; Mann-Whitney test. (F) Wright-Giemsa staining of E12.5 Gps2+/+ and Gps2−/− fetal liver cytospin preparations. Scale bars, 10 μm. (G) Wright-Giemsa staining of peripheral blood cytospins from Gps2+/+ and Gps2−/− embryos at E12.5. Scale bars, 20 μm. (H) The percentage of enucleated red blood cells in peripheral blood cytospins from Gps2+/+ and Gps2−/− embryos at E12.5 (n = 3/group, mean ± SEM). Two-tailed unpaired t test. (I) Representative flow cytometry profiles of R1 to R5 erythroblast populations labeled with CD71 and Ter119 in fetal liver from Gps2+/+ embryos at E12.5. (J) The frequency of R1 to R5 cells in Gps2+/+ (n = 5), Gps2+/− (n = 8), and Gps2−/− (n = 4) E12.5 fetal livers. Data are presented as mean ± SEM; 2-tailed unpaired t test. (K) Representative flow cytograms of Gps2+/+ Ter119hi fetal liver cells (E12.5) separated into 3 populations (S1, S2, and S3) based on the forward light scatter (FSC) profile. (L) The frequency of S1 to S3 cells in Ter119hi fetal liver cells from each embryo at E12.5 (Gps2+/+, n = 5; Gps2+/−, n = 8; Gps2−/−, n = 4). Data are presented as mean ± SEM; 2-tailed unpaired t test. (M) Representative flow cytometry of enucleated cells in the S1 to S3 populations using Cyto-16 for nuclei and 7-aminoactinomycin D (7-AAD) for cell viability. (N) Percentages of enucleated cells in S1, S2, and S3 populations (Gps2+/+, n = 6; Gps2+/−, n = 11; Gps2−/−, n = 6). Data are presented as mean ± SEM; 2-tailed unpaired t test. (O) Real-time PCR analysis of the indicated erythroid genes in FACS-sorted CD71+Ter119+ cells from Gps2+/+ and Gps2−/− E12.5 fetal liver. Values are mean ± SEM (n = 3 replicates); 2-tailed unpaired t test. (P) Quantification of BFU-E, CFU-GM, and CFU-GEMM colonies from Methocult cultures of 4 × 104Gps2+/+, Gps2+/−, and Gps2−/− E12.5 fetal liver cells (n = 4/group). Values are mean ± SEM. *P < .05, **P < .01, ***P < .001; ns, not significant.
Next, we investigated whether deficiency of GPS2 affects erythroid terminal differentiation using flow cytometry with CD71 and Ter119 markers, which readily distinguishes various stages of erythroid differentiation (R1-R5).32 GPS2 knockout led to substantial increases in R1 and R2 cells and marked decreases in R3-R5 erythroid cells (Figure 3I-J), indicating a defect at late stages of erythroblast maturation. To further characterize terminal differentiation, Ter119hi cells were separated into 3 populations using forward light scatter and CD71, S1 (large, nucleated cells), S2 (medium, early enucleating cells), and S3 (small, enucleated cells), indicative of progressively more mature cells.33 Consistent with a late block in erythroid terminal differentiation, Gps2−/− fetal livers showed an increased proportion of S2 cells and a decreased proportion of S3 cells (Figure 3K-L). Nuclear staining analysis of the erythroblast enucleation in S1 to S3 populations showed that most enucleation events occurred in the S3 population, and deficiency of GPS2 dramatically reduced enucleation efficiency in this population (Figure 3M-N). Accordingly, the expression of certain erythroid genes, including Hbb-b1, Alas2, and Ermap, was significantly decreased in Gps2−/− CD71+Ter119+ erythroid cells from E12.5 fetal liver (Figure 3O). Notably, deletion of GPS2 had no effects on the number of the BFU-Es, CFU-GMs, and CFU-GEMMs cultured from E12.5 fetal liver (Figure 3P); moreover, analysis of HSPCs in E12.5 fetal livers revealed no reduction in the fraction of Lin−Sca1+c-Kit+ cells (LSKs), CMPs, GMPs, and MEPs in Gps2−/− fetal livers (supplemental Figure 7A-E), indicating that the progenitor cell compartment is not affected in Gps2−/− fetal livers. Taken together, these results indicate that deficiency of GPS2 blocks terminal erythroid differentiation.
Our previous results showed that GPS2-silenced UCB CD34+ cells might have a disadvantage during in vivo repopulation. We thus examined the effect of GPS2 ablation on the hematopoietic repopulating ability of mouse fetal liver. Engraftment analysis at 6 weeks after transplantation showed that the repopulation rate of Gps2+/+ and Gps2−/− fetal liver cells is 28/30 and 2/21, respectively. Both reconstituted recipients transplanted with Gps2−/− fetal liver cells showed <5% donor-derived cells. These data suggest that GPS2 deficiency remarkably decreases the hematopoietic repopulating ability of mouse fetal liver.
GPS2 enhances EKLF-mediated transcriptional activity
Considering that GATA1 and EKLF are 2 major transcriptional regulators of erythropoiesis and the erythroid differentiation defects caused by GPS2 deficiency are similar to those caused by GATA1 and EKLF deficiency,2,34 we hypothesized that GPS2 might regulate erythroid differentiation through modifying the transcriptional activity of GATA1 or EKLF. ALAS2 is an erythroid-specific protein and an important rate-limiting enzyme in heme synthesis, which is regulated by both GATA1 and EKLF.35 We thus used a luciferase reporter gene driven by ALAS2 promoter to examine the effect of GPS2 on GATA1‐ and EKLF-mediated transcriptional activity. The results showed that overexpression of GPS2 significantly enhanced EKLF-mediated transcriptional activity in a dose‐dependent manner but had no obvious effect on GATA1-mediated transcriptional activity (Figure 4A-B). Both AHSP and HBB are typical target genes of EKLF.4 Experiments using an AHSP or HBB promoter‐driven luciferase reporter construct also yielded similar results (Figure 4C-D). These findings indicate that GPS2 specifically regulates the transcription activity of EKLF, but not GATA1. In addition, disruption of the EKLF-binding site in the ALAS2 promoter driven–luciferase reporter construct obviously weakened the promotion of EKLF transcriptional activity by GPS2 overexpression (Figure 4E), suggesting that GPS2 enhances ALAS2 expression in an EKLF‐dependent manner.
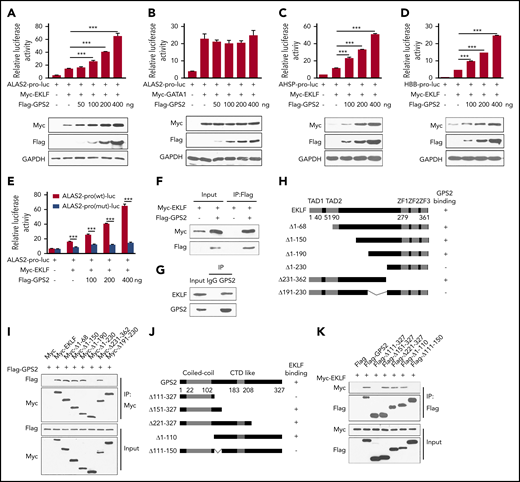
GPS2 interacts with EKLF and enhances EKLF-mediated transcriptional activity. (A-E) Luciferase reporter assays to test the effects of GPS2 on EKLF- and GATA1-mediated transcriptional activity. HEK293T cells were transfected with various combinations of plasmids as indicated below each column. Relative luciferase activity was measured after 48 hours by a dual-luciferase reporter assay. All activities were normalized to pGL3-Basic activity. Overexpression of GPS2 promotes EKLF-induced (A), but not GATA1-induced (B), luciferase activity of ALAS2-pro-luc plasmid and increases EKLF-induced luciferase activity of AHSP-pro-luc (C) and HBB-pro-luc plasmid (D). Disruption of the EKLF-binding site in the ALAS2-pro-luc reporter construct obviously weakened the promotion of EKLF-induced luciferase activity by GPS2 overexpression (E). All values are mean ± SEM (n = 3 replicates). (F) HEK293T cells were transfected with Myc-EKLF together with Flag-GPS2 or control plasmid. Immunoblot analysis of Myc- and Flag-tagged proteins in cell lysates immunoprecipitated (IP) with anti-Flag M2 agarose. (G) Lysates of MEL cells were immunoprecipitated with GPS2 antibody or normal rabbit immunoglobulin G (IgG) and then subjected to immunoblot with EKLF or GPS2 antibody. (H) A schematic representation of EKLF WT and deletion mutants. (I) HEK293T cells were transfected with various combinations of plasmids encoding Flag-GPS2 and Myc-EKLF or EKLF deletion mutants as indicated. Cell lysates were pulled down with anti-c-Myc agarose and subjected to immunoblot with Myc or Flag antibody. (J) A schematic representation of GPS2 WT and deletion mutants. (K) HEK293T cells were transfected with various combinations of plasmids encoding Myc-EKLF and Flag-GPS2 or GPS2 deletion mutants as indicated. Cell lysates were pulled down with anti-Flag M2 agarose and subjected to immunoblot with Myc or Flag antibody. Data are representative of three independent experiments (F-G,I,K). ***P < .001; 2-tailed unpaired t test.
GPS2 interacts with EKLF and enhances EKLF-mediated transcriptional activity. (A-E) Luciferase reporter assays to test the effects of GPS2 on EKLF- and GATA1-mediated transcriptional activity. HEK293T cells were transfected with various combinations of plasmids as indicated below each column. Relative luciferase activity was measured after 48 hours by a dual-luciferase reporter assay. All activities were normalized to pGL3-Basic activity. Overexpression of GPS2 promotes EKLF-induced (A), but not GATA1-induced (B), luciferase activity of ALAS2-pro-luc plasmid and increases EKLF-induced luciferase activity of AHSP-pro-luc (C) and HBB-pro-luc plasmid (D). Disruption of the EKLF-binding site in the ALAS2-pro-luc reporter construct obviously weakened the promotion of EKLF-induced luciferase activity by GPS2 overexpression (E). All values are mean ± SEM (n = 3 replicates). (F) HEK293T cells were transfected with Myc-EKLF together with Flag-GPS2 or control plasmid. Immunoblot analysis of Myc- and Flag-tagged proteins in cell lysates immunoprecipitated (IP) with anti-Flag M2 agarose. (G) Lysates of MEL cells were immunoprecipitated with GPS2 antibody or normal rabbit immunoglobulin G (IgG) and then subjected to immunoblot with EKLF or GPS2 antibody. (H) A schematic representation of EKLF WT and deletion mutants. (I) HEK293T cells were transfected with various combinations of plasmids encoding Flag-GPS2 and Myc-EKLF or EKLF deletion mutants as indicated. Cell lysates were pulled down with anti-c-Myc agarose and subjected to immunoblot with Myc or Flag antibody. (J) A schematic representation of GPS2 WT and deletion mutants. (K) HEK293T cells were transfected with various combinations of plasmids encoding Myc-EKLF and Flag-GPS2 or GPS2 deletion mutants as indicated. Cell lysates were pulled down with anti-Flag M2 agarose and subjected to immunoblot with Myc or Flag antibody. Data are representative of three independent experiments (F-G,I,K). ***P < .001; 2-tailed unpaired t test.
GPS2 interacts with EKLF
We next examined the interaction between GPS2 and EKLF. Coimmunoprecipitation assays in HEK293T cells showed that GPS2 bound to EKLF in vitro (Figure 4F). Moreover, the endogenous interaction between GPS2 and EKLF was confirmed in mouse erythroleukemia (MEL) cells (Figure 4G). Mapping of the binding domain targeted by GPS2 revealed that deletion of amino acids (aa) 191-230 blocked the interaction between GPS2 and EKLF, indicating that this middle region is essential for EKLF binding to GPS2 (Figure 4H-I; supplemental Figure 8A). In addition, the EKLF-interacting surface of GPS2 was mapped to aa 111-150 (Figure 4J-K; supplemental Figure 8B).
GPS2 increases protein stability of EKLF but does not alter its ubiquitination
We noticed that GPS2 dose-dependently increased EKLF protein level in luciferase reporter gene assays, suggesting that GPS2 may regulate the protein stability of EKLF (Figure 4A-D). To confirm, we demonstrated decreased protein levels but unchanged mRNA levels of EKLF in GPS2-knockdown MEL cells and in Gps2−/− fetal liver (Figure 5A-D). We next carried out the CHX chase experiment in HEK293T cells and found that coexpression of Flag-GPS2 with Myc-EKLF led to increased half-life of EKLF protein (Figure 5E). In addition, GPS2 overexpression also increased the half-life of endogenous EKLF protein in MEL cells, while knockdown of GPS2 accelerated the degradation of EKLF protein (Figure 5F-G). We further demonstrated that GPS2-Δ111-150 mutant lacking the EKLF-binding domain was unable to enhance EKLF protein stability (Figure 5H), and WT GPS2 failed to stabilize EKLF-Δ191-230 mutant lacking the GPS2-binding domain (Figure 5I). Although earlier studies have shown that deletion of the EKLF TAD1, PEST1, or PEST2 domain retards EKLF degradation, GPS2 still enhanced the protein stability of the EKLF-Δ1-68 mutant lacking all 3 of these domains (Figure 5J). Thus, GPS2 stabilizes EKLF protein in a manner dependent on the interaction of GPS2 with EKLF but independent of the EKLF TAD1 and PEST domains.

GPS2 increases protein stability of EKLF but does not alter its ubiquitination. (A-B) Immunoblot analysis of GPS2 and EKLF protein levels in GPS2-knockdown and control MEL cells (A), as well as in Gps2+/+ and Gps2−/− E12.5 fetal livers (B). (C-D) Real-time PCR analysis of GPS2 and EKLF mRNA levels in GPS2-knockdown and control MEL cells (C), as well as in Gps2+/+ and Gps2−/− E12.5 fetal livers (D). (E) HEK293T cells transfected with Myc-EKLF together with Flag-GPS2 or control vector were treated with CHX for indicated times and then analyzed by western blot. (F-G) MEL cells with/without stable overexpression of GPS2 (F) or knockdown of GPS2 (G) were treated with CHX for indicated times and then analyzed by western blot. (H) HEK293T cells transfected with Myc-EKLF together with Flag-GPS2-Δ111-150 or control vector were treated with CHX for indicated times and then analyzed by western blot. (I) HEK293T cells transfected with Myc-EKLF-Δ191-230 together with Flag-GPS2 or control vector were treated with CHX for indicated times and then analyzed by immunoblot. (J) HEK293T cells transfected with Myc-EKLF-Δ1-68 together with Flag-GPS2 or control vector were treated with CHX for indicated times and then analyzed by western blot. Representative western blot and quantification of relative protein levels are shown. (K) HEK293T cells transfected with Myc-EKLF together with Flag-GPS2 or control vector were treated with vehicle, MG132 (20 μM), or E64 (50 μM) for 6 hours. Cell lysates were subjected to immunoblot with Myc or Flag antibody. (L) HEK293T cells transfected with various combinations of plasmids as indicated below each column were left untreated or treated with MG132 for 6 hours. The relative luciferase activities were measured after 48 hours by a dual-luciferase reporter assay. All activities were normalized to pGL3-Basic activity. (M) HEK293T cells were transfected with various combinations (above lanes) of plasmids encoding Myc-EKLF, Flag-GPS2, and hemagglutinin-ubiquitin (HA-Ub). Before collection, cells were treated with MG132 (20 μM) for 6 hours. Immunoblot analysis of EKLF ubiquitination (detected by EKLF antibody) in cell lysates immunoprecipitated with HA antibody. (N) MEL cells with or without stable GPS2 knockdown were pretreated with MG132 (20 μM) for 6 hours before collection. The cell lysates were immunoprecipitated with tandem ubiquitin binding entities (TUBEs)-conjugated agarose beads and then subjected to immunoblot analysis of EKLF ubiquitination. Data are representative of 3 independent experiments (A-B,K,M-N). All values are mean ± SEM (n = 3 replicates). *P < .05, **P < .01, ***P < .001; 2-tailed unpaired t test (C-D,L) or 2-way ANOVA test (E-J).
GPS2 increases protein stability of EKLF but does not alter its ubiquitination. (A-B) Immunoblot analysis of GPS2 and EKLF protein levels in GPS2-knockdown and control MEL cells (A), as well as in Gps2+/+ and Gps2−/− E12.5 fetal livers (B). (C-D) Real-time PCR analysis of GPS2 and EKLF mRNA levels in GPS2-knockdown and control MEL cells (C), as well as in Gps2+/+ and Gps2−/− E12.5 fetal livers (D). (E) HEK293T cells transfected with Myc-EKLF together with Flag-GPS2 or control vector were treated with CHX for indicated times and then analyzed by western blot. (F-G) MEL cells with/without stable overexpression of GPS2 (F) or knockdown of GPS2 (G) were treated with CHX for indicated times and then analyzed by western blot. (H) HEK293T cells transfected with Myc-EKLF together with Flag-GPS2-Δ111-150 or control vector were treated with CHX for indicated times and then analyzed by western blot. (I) HEK293T cells transfected with Myc-EKLF-Δ191-230 together with Flag-GPS2 or control vector were treated with CHX for indicated times and then analyzed by immunoblot. (J) HEK293T cells transfected with Myc-EKLF-Δ1-68 together with Flag-GPS2 or control vector were treated with CHX for indicated times and then analyzed by western blot. Representative western blot and quantification of relative protein levels are shown. (K) HEK293T cells transfected with Myc-EKLF together with Flag-GPS2 or control vector were treated with vehicle, MG132 (20 μM), or E64 (50 μM) for 6 hours. Cell lysates were subjected to immunoblot with Myc or Flag antibody. (L) HEK293T cells transfected with various combinations of plasmids as indicated below each column were left untreated or treated with MG132 for 6 hours. The relative luciferase activities were measured after 48 hours by a dual-luciferase reporter assay. All activities were normalized to pGL3-Basic activity. (M) HEK293T cells were transfected with various combinations (above lanes) of plasmids encoding Myc-EKLF, Flag-GPS2, and hemagglutinin-ubiquitin (HA-Ub). Before collection, cells were treated with MG132 (20 μM) for 6 hours. Immunoblot analysis of EKLF ubiquitination (detected by EKLF antibody) in cell lysates immunoprecipitated with HA antibody. (N) MEL cells with or without stable GPS2 knockdown were pretreated with MG132 (20 μM) for 6 hours before collection. The cell lysates were immunoprecipitated with tandem ubiquitin binding entities (TUBEs)-conjugated agarose beads and then subjected to immunoblot analysis of EKLF ubiquitination. Data are representative of 3 independent experiments (A-B,K,M-N). All values are mean ± SEM (n = 3 replicates). *P < .05, **P < .01, ***P < .001; 2-tailed unpaired t test (C-D,L) or 2-way ANOVA test (E-J).
In line with the previous report that EKLF degradation is mediated by the proteasome pathway,5 we observed a significantly increased EKLF protein level by treatment with the proteasome inhibitor MG132, but not the lysosomal inhibitor E64, in HEK293T cells transfected with Myc-EKLF (Figure 5K). In MG132-treated HEK293T cells, GPS2 had only mild effects on the stability of EKLF protein and EKLF-mediated luciferase expression driven by HBB promoter (Figure 5K-L), indicating that GPS2 promotes EKLF-mediated transcriptional activity by preventing proteasome-mediated EKLF protein degradation. We next examined whether GPS2 affects EKLF polyubiquitination and found that neither overexpression of GPS2 in HEK293T cells nor knockdown of GPS2 in MEL cells had an obvious effect on the total ubiquitination of EKLF (Figure 5M-N). GPS2 has been reported to inhibit Ubc13, an E2 that works with many E3 ligases and mediates K63-linked ubiquitination, which could potentially regulate the stability of a plethora of proteins. We thus investigated whether GPS2 stabilizes EKLF depending on Ubc13. In HEK293T cells, overexpression of Ubc13 did not affect the protein level of EKLF or K63-linked ubiquitination of EKLF (supplemental Figure 9A-B). Consistently, MEL cells treated with the Ubc13 inhibitor NSC697923 showed an unaltered half-life of EKLF protein (supplemental Figure 9C). Thus, Ubc13 does not affect the protein stability and K63-linked ubiquitination of EKLF. Moreover, neither overexpression of GPS2 in HEK293T cells nor knockdown of GPS2 in MEL cells affected the K63-linked ubiquitination of EKLF (supplemental Figure 9D-E). Taken together, these results show that GPS2 inhibits EKLF degradation through proteasomes but does not affect EKLF polyubiquitination.
Identification of aa 191-230 region is important for EKLF stability
By multiple amino acid sequence alignment, we found that the aa 191-230 region of EKLF is conserved in mammal specials (Figure 6A), indicating that this region may have a role in maintaining the structure or function of EKLF. Indeed, we found that the stability of EKLF-Δ191-230 mutant was decreased compared with WT EKLF (Figure 6B), but its ubiquitination was not substantially reduced (Figure 6C). Accordingly, deletion of aa 191-230 resulted in a reduction of EKLF-mediated transcriptional activity (Figure 6D). Notably, various mutations have been found in the aa 191-230 region of EKLF (supplemental Table 1). Among them, the c.632A>G (p.Gln211Arg) mutation has been identified to be associated with increased fetal hemoglobin levels, and the stability of EKLF protein with this mutation was predicted to be decreased.36 We demonstrated that the stability of EKLF (Gln211Arg) mutant was decreased compared with WT EKLF (Figure 6E). Accordingly, Gln211Arg mutation weakened the interaction with GPS2 (Figure 6F), and GPS2 had a mild promotive effect on the stability of EKLF (Gln211Arg) mutant compared with WT EKLF protein (Figure 6G). These data reveal that the aa 191-230 region is important for EKLF stability.
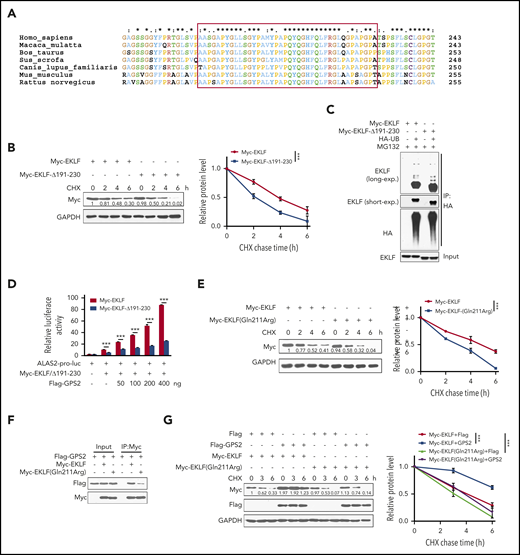
Identification of the aa 191-230 region is important for EKLF stability. (A) Multiple amino acid sequence alignment of the aa 191-230 region (in red box) on EKLF proteins from the indicated species using the Clustal X 2.0 program. (B) HEK293T cells transfected with Myc-EKLF or Myc-EKLF-Δ191-230 were treated with CHX for indicated times and then analyzed by western blot. Representative western blot and quantification of relative protein levels are shown. (C) HEK293T cells were transfected with various combinations (above lanes) of plasmids encoding Myc-EKLF, Myc-EKLF-Δ191-230, and HA-Ub. Before collection, cells were treated with MG132 (20 μM) for 6 hours. Immunoblot analysis of EKLF ubiquitination (detected by EKLF antibody) in cell lysates immunoprecipitated with HA antibody. (D) HEK293T cells were transfected with various combinations of plasmids as indicated below each column. The relative luciferase activities were measured after 48 hours by a dual-luciferase reporter assay. All activities were normalized to pGL3-Basic activity. (E) HEK293T cells transfected with Myc-EKLF or Myc-EKLF (Gln211Arg) were treated with CHX for indicated times and then analyzed by western blot. Representative western blot and quantification of relative protein levels are shown. (F) HEK293T cells were transfected with Flag-GPS2 together with Myc-EKLF or Myc-EKLF (Gln211Arg). Cell lysates were pulled down with anti-c-Myc agarose and subjected to immunoblot with Myc or Flag antibody. (G) HEK293T cells transfected with Myc-EKLF or Myc-EKLF (Gln211Arg) together with Flag-GPS2 or control vector were treated with CHX for indicated times and then analyzed by western blot. Representative western blot and quantification of relative protein levels are shown. Data are representative of 3 independent experiments (C,F). Values are mean ± SEM (n = 3 replicates). ***P < .001; 2-tailed unpaired t test (D) or 2-way ANOVA test (B,E,G).
Identification of the aa 191-230 region is important for EKLF stability. (A) Multiple amino acid sequence alignment of the aa 191-230 region (in red box) on EKLF proteins from the indicated species using the Clustal X 2.0 program. (B) HEK293T cells transfected with Myc-EKLF or Myc-EKLF-Δ191-230 were treated with CHX for indicated times and then analyzed by western blot. Representative western blot and quantification of relative protein levels are shown. (C) HEK293T cells were transfected with various combinations (above lanes) of plasmids encoding Myc-EKLF, Myc-EKLF-Δ191-230, and HA-Ub. Before collection, cells were treated with MG132 (20 μM) for 6 hours. Immunoblot analysis of EKLF ubiquitination (detected by EKLF antibody) in cell lysates immunoprecipitated with HA antibody. (D) HEK293T cells were transfected with various combinations of plasmids as indicated below each column. The relative luciferase activities were measured after 48 hours by a dual-luciferase reporter assay. All activities were normalized to pGL3-Basic activity. (E) HEK293T cells transfected with Myc-EKLF or Myc-EKLF (Gln211Arg) were treated with CHX for indicated times and then analyzed by western blot. Representative western blot and quantification of relative protein levels are shown. (F) HEK293T cells were transfected with Flag-GPS2 together with Myc-EKLF or Myc-EKLF (Gln211Arg). Cell lysates were pulled down with anti-c-Myc agarose and subjected to immunoblot with Myc or Flag antibody. (G) HEK293T cells transfected with Myc-EKLF or Myc-EKLF (Gln211Arg) together with Flag-GPS2 or control vector were treated with CHX for indicated times and then analyzed by western blot. Representative western blot and quantification of relative protein levels are shown. Data are representative of 3 independent experiments (C,F). Values are mean ± SEM (n = 3 replicates). ***P < .001; 2-tailed unpaired t test (D) or 2-way ANOVA test (B,E,G).
GPS2 promotes erythroid cell differentiation depending on EKLF
We next investigated the function of GPS2/EKLF interaction in erythroid differentiation. Overexpression of the WT GPS2 in MEL cells significantly increased the proportion of benzidine-positive cells induced by hexamethylene bisacetamide (HMBA), as well as the expression of the EKLF target genes Ahsp and Hbb-b1 (Figure 7A-D). In contrast, the GPS2-Δ111-150 mutant that lacks the EKLF-binding domain was unable to accelerate the erythroid differentiation of MEL cells (Figure 7A-D). Interestingly, the GPS2-Δ1-110 mutant containing the EKLF‐binding domain but lacking the NCOR1-binding domain showed a clear promotive effect on erythroid differentiation similar to WT GPS2 (Figure 7A-D).9 Consistent with these results, overexpression of GPS2 and GPS2-Δ1-110 mutant, but not GPS2-Δ111-150 mutant, promoted erythroid differentiation in human UCB CD34+ cells (Figure 7E; supplemental Figure 10A-B). Our findings support an NCOR1-independent role of GPS2 in erythroid differentiation. The results that knockdown of NCOR1 had no significant effect on erythroid differentiation of UCB CD34+ cells further supported this conclusion (Figure 7F-G; supplemental Figure 11A-C).

GPS2 promotes erythroid cell differentiation depending on EKLF. (A-D) Control cells and MEL cells with stable overexpression of WT GPS2 or GPS2 deletion mutants were left untreated or treated with HMBA for indicated times. Immunoblot analysis of GPS2 and EKLF protein levels (A). Hemoglobin production was analyzed by benzidine staining at days 3 and 4. Representative images of benzidine-stained cells are shown; scale bar, 20 μm (B). The percentage of benzidine-positive cells was calculated (C). Real-time PCR analysis of Ahsp and Hbb-b1 mRNA levels at day 3 (D). (E) CD34+GFP+ cells were selected from human UCB CD34+ cells infected with lentivirus expressing WT GPS2 or GPS2 deletion mutants and cultured in the erythroid differentiation medium for the indicated times. The proportion of CD71+GPA+ cells at days 0, 3, and 5 was analyzed by flow cytometry. (F-G) Human UCB CD34+ cells infected with NCOR1 or control shRNA lentivirus were cultured in the erythroid differentiation medium for the indicated times. The proportion of CD71+GPA+ cells at days 0, 3, and 5 was analyzed by flow cytometry (F). mRNA levels of the indicated erythroid genes in FACS-isolated CD71+GPA+ cells at day 3 were analyzed with real-time PCR (G). (H-L) Control cells and MEL cells with stable GPS2 knockdown or GPS2 knockdown and EKLF overexpression were left untreated or treated with HMBA for the indicated times. EKLF and GPS2 protein levels were analyzed by western blot (H). A representative photograph of the color of cell pellets at day 4 (I). Hemoglobin production was analyzed by benzidine staining at days 4 and 5. Representative images of benzidine-stained cells are shown; scale bar, 20 μm (J). The percentage of benzidine-positive cells was calculated (K). Relative mRNA levels of Alas2, Ahsp, and Hbb-b1 at day 3 were analyzed by real-time PCR (L). (M) CD34+GFP+RFP+ cells were selected from human UCB CD34+ cells infected with GPS2 shRNA lentivirus and EKLF overexpression or control lentivirus and then cultured in erythroid differentiation medium for the indicated times. The proportion of CD71+GPA+ cells at day 0, 3, and 5 was analyzed by flow cytometry. All values are mean ± SEM (n = 3 replicates). *P < .05, **P < .01, ***P < .001; 2-tailed unpaired t test.
GPS2 promotes erythroid cell differentiation depending on EKLF. (A-D) Control cells and MEL cells with stable overexpression of WT GPS2 or GPS2 deletion mutants were left untreated or treated with HMBA for indicated times. Immunoblot analysis of GPS2 and EKLF protein levels (A). Hemoglobin production was analyzed by benzidine staining at days 3 and 4. Representative images of benzidine-stained cells are shown; scale bar, 20 μm (B). The percentage of benzidine-positive cells was calculated (C). Real-time PCR analysis of Ahsp and Hbb-b1 mRNA levels at day 3 (D). (E) CD34+GFP+ cells were selected from human UCB CD34+ cells infected with lentivirus expressing WT GPS2 or GPS2 deletion mutants and cultured in the erythroid differentiation medium for the indicated times. The proportion of CD71+GPA+ cells at days 0, 3, and 5 was analyzed by flow cytometry. (F-G) Human UCB CD34+ cells infected with NCOR1 or control shRNA lentivirus were cultured in the erythroid differentiation medium for the indicated times. The proportion of CD71+GPA+ cells at days 0, 3, and 5 was analyzed by flow cytometry (F). mRNA levels of the indicated erythroid genes in FACS-isolated CD71+GPA+ cells at day 3 were analyzed with real-time PCR (G). (H-L) Control cells and MEL cells with stable GPS2 knockdown or GPS2 knockdown and EKLF overexpression were left untreated or treated with HMBA for the indicated times. EKLF and GPS2 protein levels were analyzed by western blot (H). A representative photograph of the color of cell pellets at day 4 (I). Hemoglobin production was analyzed by benzidine staining at days 4 and 5. Representative images of benzidine-stained cells are shown; scale bar, 20 μm (J). The percentage of benzidine-positive cells was calculated (K). Relative mRNA levels of Alas2, Ahsp, and Hbb-b1 at day 3 were analyzed by real-time PCR (L). (M) CD34+GFP+RFP+ cells were selected from human UCB CD34+ cells infected with GPS2 shRNA lentivirus and EKLF overexpression or control lentivirus and then cultured in erythroid differentiation medium for the indicated times. The proportion of CD71+GPA+ cells at day 0, 3, and 5 was analyzed by flow cytometry. All values are mean ± SEM (n = 3 replicates). *P < .05, **P < .01, ***P < .001; 2-tailed unpaired t test.
We further examined whether there is a direct relationship between EKLF downregulation and erythroid differentiation in GPS2-knockdown cells. In MEL cells, knockdown of GPS2 led to a remarkable decrease in EKLF protein level (Figure 7H). A dark-red cell pellet was observed in control shRNA–transduced MEL cells induced by HMBA, while a pale cell pellet was observed in GPS2 shRNA–transduced MEL cells, indicating a marked reduction in hemoglobin production (Figure 7I). Accordingly, GPS2-knockdown MEL cells had fewer benzidine-positive cells and decreased mRNA levels of Alas2, Ahsp, and Hbb-b1 after HMBA treatment (Figure 7J-L). We then re-expressed EKLF in GPS2-knockdown MEL cells by infecting EKLF overexpression lentivirus (Figure 7H). As expected, re-expression of EKLF completely rescued the inhibited erythroid differentiation in GPS2-knockdown MEL cells (Figure 7I-L). We also observed that EKLF reversed GPS2-knockdown–mediated blockage of erythroid differentiation in human UCB CD34+ cells (Figure 7M; supplemental Figure 12A-B). These data confirm that GPS2 promotes erythroid cell differentiation depending on EKLF.
Discussion
Using a systematic approach, we uncovered a unique and fundamental role of GPS2 in erythrocyte differentiation. We found that GPS2 expression was upregulated during the erythroid differentiation of human UCB CD34+ cells induced by EPO. Knockdown of GPS2 in human CD34+ cells suppressed erythroid differentiation in vitro, as shown by the decreased proportion of GPA+ cells and benzidine-positive cells as well as the reduced expression of certain erythroid genes. GPS2-silenced human CD34+ cells transplanted into NSG mice displayed impaired erythropoiesis in vivo. Moreover, mice embryos lacking GPS2 developed severe anemia owing to defects in late-stage erythropoiesis in fetal liver. Thus, we have elucidated for the first time a previously unknown role of GPS2 in erythroid differentiation, particularly erythroblast maturation.
Our study identified a novel nontranscriptional role of GPS2 and specifically linked GPS2 with the modulation of EKLF protein stability. EKLF has been reported to be degraded through a ubiquitin-dependent proteasome pathway, whereas we found that only a small portion of EKLF protein was shown to be covalently modified by polyubiquitination, which is in line with the previous results.5 We further demonstrated that most EKLF protein noncovalently interacted with ubiquitin, which is supported by the fact that EKLF TAD1 domain is able to form noncovalent interactions with ubiquitin and contributes to ubiquitin-mediated degradation of EKLF.6 However, we failed to detect a significant effect of GPS2 on EKLF ubiquitination. Moreover, although EKLF-Δ191-230 mutant had increased instability compared with WT EKLF, it showed unaltered ubiquitination. These results indicated that GPS2 promotes EKLF stability in a ubiquitin-independent manner. Substrate proteins should be recognized, unfolded, and deubiquitinated by the 19S regulatory particle before being funneled into the proteolytic core.37 We thus proposed that GPS2 may prevent ubiquitinated EKLF from entering into the proteasome, thereby decrease degradation of EKLF. However, the underlying mechanism is remained to be revealed by further studies. This new role of GPS2 in protein stability regulation is distinct from previously reported functions of GPS2 relying on the direct inhibition of Ubc13 enzymatic activity.19,20 Our findings therefore raise the possibility that GPS2 performs its nontranscriptional function in distinct biological processes by different mechanisms.
It is clear that NCOR1 plays an essential role in erythrocyte differentiation. NCOR1-null mice are embryonically lethal, and the majority of Ncor1−/− embryos appear to die of anemia by E15.5 owing to defects in definitive erythropoiesis.24 In addition, knockdown of NCOR1 impairs the erythroid differentiation of K562 cells.25,27 As GPS2 is a main component of the NCOR1-HDAC3 complex,38 it is easy to think that GPS2’s function in erythropoiesis may depend on NCOR1. However, previous results showed that neither NCOR1 knockdown nor HDAC3 inhibition can suppress erythroid differentiation in MEL cells.26 We found that silence of GPS2 significantly inhibits HMBA-induced erythroid differentiation in MEL cells. The GPS2-Δ1-110 mutant lacking the NCOR1-binding domain has promotive effects comparable to WT GPS2 on erythroid differentiation in MEL cells and human UCB CD34+ cells. In addition, knockdown of NCOR1 in human UCB CD34+ cells has no effect on erythroid differentiation. These findings support an NCOR1-independent role of GPS2 in erythroid differentiation.
The lethality of Gps2−/− mice on C57BL/6N background became apparent at E10.5 and completed by E14.5, while this early onset of lethality was delayed to E13.5 after crossing with 129S2/Sv mice. However, EKLF-knockout mice die at E14, which is later than Gps2−/− mice, suggesting that there are EKLF-independent functions of GPS2 at the early stage of mouse embryonic development. We found that knockdown of GPS2 led to a trend toward lower repopulation of human UCB CD34+ cells in NSG mice, and GPS2-deficient mouse fetal liver almost completely lost its hematopoietic reconstitution ability, indicating an important role of GPS2 in HSC repopulation. As EKLF is not essential for nonerythroid lineage reconstitution,39 GPS2’s function in HSC repopulation may be independent of EKLF. GPS2 is a multifunctional protein that can act as a subunit of the NCoR/silencing mediator of retinoic acid and thyroid hormone receptor corepressor complexes, inhibit Ubc13, and regulate mitochondria biogenesis and function.9,15,40 Any of these pathways could contribute to the defects in hematopoietic repopulation seen in GPS2 deficiency.41-44 Otherwise, it is also possible that the early embryonic lethality of GPS2-knockout mice may be due to defects in the development of other nonhematopoietic tissues at the early stage of embryonic development. However, the precise regulation mechanism of GPS2 in hematopoietic repopulation and the possible nonhematopoietic functions of GPS2 remain to be determined in further studies.
Certain mutations in the human KLF1 gene have been associated with severe hematologic disorders.45,46 In addition, several benign hematologic conditions are due to KLF1 haploinsufficiency.3 A few mutations leading to KLF1 insufficiency are located in the linker between the transactivation domains and DNA-binding domains of KLF1; however, the mechanisms are still unknown.47 We demonstrated that aa 191-230 in EKLF protein are essential for GPS2-mediated stabilization of EKLF. Interestingly, this region is conserved in mammals by multiple sequence alignment analysis, and various mutants are present in this region according to the National Center for Biotechnology Information dbSNP database. Among those mutations, the c.632A>G (p.Gln211Arg) mutation is associated with increased fetal hemoglobin levels, and the c.604G>A (p.Gly202Arg) mutation is responsible for the In(Lu) phenotype.36,48 However, the underlying mechanism is still unclear. Our results show that GPS2 cannot effectively protect EKLF-Δ191-230 and the EKLF (Gln211Arg) mutant from proteasome-mediated degradation, which brings new insight into the understanding of hematologic disorders caused by KLF1 insufficiency.
For original data, please contact either xiaomingyang@sina.com or yrh1980110@126.com.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Xue-Ming Zhang (National Center of Biomedical Analysis, Beijing, China) for providing hemagglutinin-ubiquitin plasmids.
This work was supported by grants from the National Natural Science Foundation of China (81600081) and the State Key Laboratory of Proteomics (SKLP-K201404, SKLP-O201413).
Authorship
Contribution: W.-B.M. contributed all experiments; W.-B.M. and R.-H.Y. contributed in vitro experiments, assisted by X.L., G.-M.R., W.Z., J.Z., S.-S.T., and D.-X.L.; W.-B.M., R.-H.Y., X.-H.W., and H.-H.T. performed murine in vivo experiments, assisted by J.-J.B., M.Y., C.-H.G., and C.-Y.L.; H.C. and Y.-Q.Z. contributed mice or reagents. R.-H.Y., C.-Y.L., and W.-B.M. designed the experiments; W.-B.M. and R.-H.Y. analyzed the data; X.-M.Y., R.-H.Y., and W.-B.M. wrote the manuscript; and X.-M.Y. and R.-H.Y. supervised the project.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Xiao-Ming Yang, State Key Laboratory of Proteomics, Beijing Proteome Research Center, Beijing Institute of Lifeomics, 27 Taiping Rd, Haidian District, Beijing 100850, People’s Republic of China; e-mail: xiaomingyang@sina.com; and Rong-Hua Yin, State Key Laboratory of Proteomics, Beijing Proteome Research Center, Beijing Institute of Lifeomics, 27 Taiping Rd, Haidian District, Beijing 100850, People’s Republic of China; e-mail: yrh1980110@126.com.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal