

In this issue of Blood, examine the incidence and fate of clonal hematopoiesis (CH) in 42 long-term survivors of allogeneic hematopoietic stem cell transplantation (HSCT) and their sibling donors.1
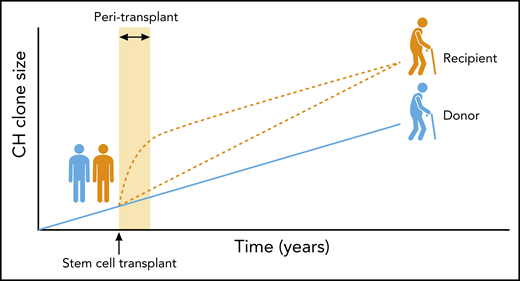
Boettcher et al report that in sibling allogeneic blood stem cell transplants, donor-derived CH expands more significantly in the recipient (orange lines) than it does in the donor (blue line), when assessed many years posttransplantation. The precise timing of the additional clonal expansion in the recipient remains unknown. One possibility is that mutant stem cells gain a transient growth advantage early after transplantation and in the context of a prevailing inflammatory environment, with growth rate reducing thereafter (upper orange line). Such behaviour has been reported in mouse models of CH. Alternatively, the recipient might continue to offer a slightly more favourable environment for the expansion of CH during and after the peri-transplant period, resulting in a persistently higher clonal growth rate (lower orange line). It is also possible that both of these alternatives operate at different times and to different extents to produce the eventual clone size observed. Future studies employing more subjects and multiple time points will be required to determine which of these scenarios prevails and the relevance to clinical outcomes. Professional illustration by Patrick Lane, ScEYEnce Studios.
Boettcher et al report that in sibling allogeneic blood stem cell transplants, donor-derived CH expands more significantly in the recipient (orange lines) than it does in the donor (blue line), when assessed many years posttransplantation. The precise timing of the additional clonal expansion in the recipient remains unknown. One possibility is that mutant stem cells gain a transient growth advantage early after transplantation and in the context of a prevailing inflammatory environment, with growth rate reducing thereafter (upper orange line). Such behaviour has been reported in mouse models of CH. Alternatively, the recipient might continue to offer a slightly more favourable environment for the expansion of CH during and after the peri-transplant period, resulting in a persistently higher clonal growth rate (lower orange line). It is also possible that both of these alternatives operate at different times and to different extents to produce the eventual clone size observed. Future studies employing more subjects and multiple time points will be required to determine which of these scenarios prevails and the relevance to clinical outcomes. Professional illustration by Patrick Lane, ScEYEnce Studios.
CH is the disproportionate clonal expansion of blood stem cells and their progeny driven by leukemia-associated somatic mutations and is associated with an increased risk of hematologic cancers and ischemic cardiovascular disease. Reassuringly, the incidence and genetic drivers of CH, in either donors or recipients, did not seem to differ from that reported for unselected individuals of similar ages. Interestingly, the study also reports on the posttransplant behavior of 5 cases of donor-engrafted CH. This scenario uniquely captures the impact of the transplant procedure and 2 different hematopoietic environments on the behavior of the same CH clone. The authors report greater clonal expansion in recipients with donor-engrafted CH compared with the growth of the same founder clone in donors, thus highlighting important features of CH biology.
There is accumulating evidence that healthy adult tissues (including blood) harbor cancer- associated somatic mutations that become ubiquitous with age.2 Although the nature of the somatic mutation is a major determinant of downstream clonal behavior, other factors are also important. Some progress has been made in identifying individuals at high risk of progressing to acute myeloid leukemia (AML),3 but the biology of CH remains poorly understood. With evidence that the inherited genome plays a relatively modest role in determining clonal behavior,4-6 there is increasing focus on nongenetic factors such as aging, proliferative stress, inflammation, and infection.7
Boettcher et al ask whether enforced HSC proliferation during allogeneic HSCT, in conjunction with an inflammatory milieu at the time of transplantation, promotes initiation, expansion, or evolution of CH in recipients compared with donors. They sequenced peripheral blood granulocyte DNA to screen for CH in 42 donor-recipient pairs. Overall, they found no difference in CH prevalence, mutation type, or clone size in donors compared with recipients, although power to detect small differences was limited by the small cohort size. Interestingly, in 5 pairs, identical mutations were detected in both donor and recipient, suggesting the transfer of preexisting CH clones. A number of informative observations stem from this and other related findings.
First, transplants from older donors were more likely to instigate (donor-derived) CH in the recipient compared with transplants from younger donors. This observation may reflect an increased frequency of stem cells with CH mutations in older donors or enhanced engraftment potential of older CH clones. In addition, older recipient age (linked to donor age in sibling transplants) may play a role by providing a more favorable environment for CH. Although CH was clearly compatible with long-term survival in this selected cohort, it is possible that larger studies may reveal that different CH mutations have distinct influences on outcomes after allogeneic HSCT, as has been observed in the autologous setting.8 The authors also identified 1 donor-recipient pair with myelodysplastic syndrome, diagnosed 18 and 21 years posttransplant, respectively, and in both cases derived from a shared founding clone. This case is informative because it reveals the long interval from mutation acquisition to malignancy and the high risk of malignant progression afforded by some mutations. The CH clone harbored a hotspot mutation in U2AF1 (among others) previously shown to confer high risk of malignant progression.3 Interestingly, the path to MDS involved acquisition of different mutations in donor vs recipient, contrasting the deterministic behavior of the clone to the stochastic process of mutation acquisition. This case may not be representative of most cases of donor-derived AML or MDS,9 but it does raise the question of whether donors should be screened for CH. Much larger studies are required to determine whether such a strategy is advisable and whether the presence of sporadic CH in donors represents a more significant risk to recipients than factors such as donor age, sex, ABO compatibility, and cytomegalovirus status.
Another important observation was the larger mutant clone size in recipients vs donors in the cases of donor-engrafted CH. This supports the premise that mutant HSCs were imparted with an additional growth advantage by peri-/posttransplant factors. Peritransplant factors include possible enrichment of harvests with or preferential engraftment of mutant HSCs and the impact of pretransplant irradiation or peritransplant inflammation or infection on CH behavior. Studies in mouse models have shown that HSCs with mutations in CH genes outcompete wild-type HSCs early after transplantation and that this can be enhanced by inflammation.10 The possibility that the recipient offers a more favorable environment for the expansion of CH beyond the peritransplant period is more difficult to investigate and would require the study of multiple time points, while being compounded by any lasting impact of peritransplant events.
Boettcher et al also provide interesting insights into the clonal architecture of CH by sequencing individual colony-forming units (CFUs), arising from single hematopoietic stem or progenitor cells, to show that CH mutations identified in bulk granulocyte DNA can be derived from a single or multiple independent clones. With 1 exception, the proportion of mutation-positive CFUs correlated well with variant allele fraction in granulocytes. In addition, the authors show that CH mutations were consistently present in myeloid cells, but were not always present in B and T cells. These findings help interpret other studies of CH that used whole blood or granulocyte DNA.
Finally, by measuring telomere length in donor and recipient CFUs, the authors demonstrate 20 years of additional hematopoietic aging in the latter. Intriguingly, within individuals with CH, telomere length was not consistently different between CFUs with and without CH mutations. The authors speculate that different mutations might have distinct requirements for telomerase activity or might activate alternative mechanisms of telomere maintenance. Furthermore, this variability may help explain differences in the risk of malignant progression associated with distinct mutations.
In the future, prospective longitudinal studies or retrospective clonal phylogenetic deconvolution will be required to build on the insights provided by the Boettcher et al study by providing more granular detail on the dynamics of CH driven by different mutations in the context of sibling and unrelated donor allogeneic HSCT. This would enhance decision making and donor choice for allogeneic HSCT and help quantify the risks associated with individual CH clones or mutations.
Conflict-of-interest disclosure: G.S.V. serves as a consultant for Kymab Ltd and OxStem Ltd. M.A.F. declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal