

Key Points
At a median follow-up of 41 months, median PFS has not been reached in previously treated CLL patients on the BTK inhibitor acalabrutinib.
Patients with previously treated CLL or SLL had favorable safety, response, and durability of response with acalabrutinib.

Abstract
Therapeutic targeting of Bruton tyrosine kinase (BTK) has dramatically improved survival outcomes for patients with chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL). Acalabrutinib is an oral, highly selective BTK inhibitor that allows for twice-daily dosing due to its selectivity. In this phase 1b/2 study, 134 patients with relapsed/refractory CLL or SLL (median age, 66 years [range, 42-85 years]; median prior therapies, 2 [range, 1-13]) received acalabrutinib 100 mg twice daily for a median of 41 months (range, 0.2-58 months). Median trough BTK occupancy at steady state was 97%. Most adverse events (AEs) were mild or moderate, and were most commonly diarrhea (52%) and headache (51%). Grade ≥3 AEs (occurring in ≥5% of patients) were neutropenia (14%), pneumonia (11%), hypertension (7%), anemia (7%), and diarrhea (5%). Atrial fibrillation and major bleeding AEs (all grades) occurred in 7% and 5% of patients, respectively. Most patients (56%) remain on treatment; the primary reasons for discontinuation were progressive disease (21%) and AEs (11%). The overall response rate, including partial response with lymphocytosis, with acalabrutinib was 94%; responses were similar regardless of genomic features (presence of del(11)(q22.3), del(17)(p13.1), complex karyotype, or immunoglobulin variable region heavy chain mutation status). Median duration of response and progression-free survival (PFS) have not been reached; the estimated 45-month PFS was 62% (95% confidence interval, 51% to 71%). BTK mutation was detected in 6 of 9 patients (67%) at relapse. This updated and expanded study confirms the efficacy, durability of response, and long-term safety of acalabrutinib, justifying its further investigation in previously untreated and treated patients with CLL/SLL. This trial was registered at www.clinicaltrials.gov as #NCT02029443.
Introduction
Chronic lymphocytic leukemia (CLL) is the most prevalent adult leukemia in the western world and typically presents at a median age of ∼70 years.1 Improved understanding of the genomic and molecular underpinnings of CLL, including the mutation status of immunoglobulin heavy variable (IGHV) genes, del(17)(p13.1), and complex karyotype, has translated into better risk stratification at diagnosis to predict time to treatment and overall survival (OS).2-11 Historically, treatment of CLL relied upon chemotherapy or chemoimmunotherapy; relapse, however, occurs even after chemoimmunotherapy.12-19 Until recently, treatment options were relatively limited, and outcomes were poor for relapsed patients. Identifying new therapeutic targets relevant for pharmacologic or immunologic interventions has been critical.
Like many other B-cell malignancies, CLL is dependent upon B-cell receptor (BCR) signaling for bisphosphate 3-kinase and Bruton tyrosine kinase (BTK).20-27 The first irreversible inhibitor of BTK, ibrutinib, demonstrated superior response, progression-free survival (PFS), and OS, compared with standard treatment approaches,28-30 and is approved for the treatment of untreated and previously treated CLL.31 Although ibrutinib has transformed outcomes for patients with CLL, it is associated with adverse events (AEs) including diarrhea, rash, atrial fibrillation, ecchymoses/bruising, major bleeding, and arthralgias/myalgias that sometimes limit its continuous use.20,28,30,32-42 The exact etiologies of these AEs are unclear, but may result from irreversible binding to alternative targets of ibrutinib, including epidermal growth factor receptor (EGFR), tyrosine-protein kinase (TEC), interleukin-2–inducible T-cell kinase (ITK), and ERBB2.43
Acalabrutinib is a highly selective, potent covalent BTK inhibitor expected to have minimal off-target activity. Acalabrutinib has rapid absorption and a short pharmacokinetic half-life with an extended pharmacodynamic response.44,45 Preclinical studies of acalabrutinib demonstrated selectivity for BTK with less inhibition of TEC and virtually no activity against EGFR (ERBB1), ERBB2, and ITK.44,45 The first-in-human study of acalabrutinib evaluated once- and twice-daily dosing and reported early efficacy and response data similar to that observed with ibrutinib.29,45 In this report, the population of patients with relapsed or refractory (R/R) CLL from that first study was expanded and follow-up was extended to examine durability of responses and long-term tolerability.
Patients and methods
This phase 1/2 multicenter study was designed to determine the recommended dose, safety, efficacy, pharmacokinetics, and pharmacodynamics of acalabrutinib in patients with R/R CLL. All patients provided written informed consent. An institutional review board approved the protocol at each clinical site. The study was conducted according to the principles of the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice.
Patients
As noted previously,45 eligibility included a diagnosis of R/R CLL/small lymphocytic lymphoma (SLL) as defined by the International Workshop on Chronic Lymphocytic Leukemia (IWCLL),46 requiring treatment per the IWCLL guidelines; ≥1 prior therapy for CLL; Eastern Cooperative Oncology Group performance status ≤2; adequate organ function; and absence of active infection. Absolute neutrophil count (ANC) ≥750/µL and platelet count ≥50 000/µL were required if no bone marrow involvement was present; no restrictions for cytopenia were applied if there was CLL bone marrow involvement. Amendments modified exclusion criteria during cohort expansion. Exclusion criteria included need for warfarin therapy or equivalent vitamin K antagonists, uncontrolled autoimmune hemolytic anemia or idiopathic thrombocytopenic purpura, or bleeding disorders (eg, history of stroke or intracranial hemorrhage within 6 months of first dose or history of bleeding diathesis). Patients with significant cardiovascular disease (uncontrolled or symptomatic arrhythmias, congestive heart failure, or myocardial infarction), any class 3 or 4 cardiac disease per New York Heart Association functional classification, left-ventricular ejection fraction ≤40%, or corrected QT interval >480 ms were excluded. History of atrial fibrillation was not an exclusion to this trial.
Evaluation and treatment
Patients underwent baseline assessments for mitogen-stimulated karyotype (n = 57); del(17)(p13.1) and del(11)(q22.3) status (n = 116); IGHV mutation status (n = 111); and β2-microglobulin (n = 101). Complex karyotype was defined as ≥3 chromosomal abnormalities. Central data were used for analysis; however, local laboratory data were used if data for del(17)(p13.1) and del(11)(q22.3) status were unavailable. Patients were successively enrolled in cohorts administering oral acalabrutinib at dosages of 100, 175, 250, and 400 mg once daily and 200 mg twice daily as part of the dose-escalation portion of the study, or 100 mg twice daily and 200 mg once daily as part of the phase 2 study. In May 2015, an amendment converted all patients to the 100-mg twice daily dose of acalabrutinib. The data cutoff for this analysis was 4 January 2019.
Assessments and end points
Patients were evaluated at screening, weekly for the first month, biweekly for the second month, monthly for the next 4 months, and every 3 months thereafter, using medical history, physical examination, and laboratory studies for signs of toxicity. After 2 years of treatment, patients were followed every 6 months. In March 2017, the protocol was amended, and patients who had completed at least 6 months of treatment were followed twice yearly; those who completed at least 3 years of treatment were followed once yearly. T-cell, natural killer cell, and monocyte numbers were measured at baseline and before cycles 3, 10, 16, 22, 28, and 34. Serum immunoglobulins were measured on the same schedule. AEs were graded according to the Common Toxicity Criteria of the National Cancer Institute, version 4.03. Study end points included overall response rate (ORR), PFS, duration of response (DOR), and safety; event-free survival (EFS) was a post hoc analysis. Response assessments, including radiologic examination, were performed at the end of cycles 2, 4, and 6, then every 6 cycles until cycle 36, and every 12 cycles thereafter. Response was evaluated based on the IWCLL criteria that includes computed tomography scan assessment,46 with incorporation of the clarification for treatment-related lymphocytosis.47
Pharmacokinetics, pharmacodynamics, and genomic profiling
Detailed pharmacokinetic analyses with a validated assay were performed during cycle 1 of therapy and were reported previously.45 BTK occupancy by acalabrutinib was measured at baseline in peripheral blood mononuclear cells (PBMCs) with a previously described drug-analog probe45 predose and 4 hours postdose on days 1 and 8 of cycle 1, and predose on day 28 of cycle 1. PBMCs were also used for T-cell profiling at predose and on day 28 of cycles 1 and 6. Additional details on the methods are provided in supplemental Methods (available on the Blood Web site).
Statistical analysis
All enrolled patients who received ≥1 dose of study drug were evaluated for safety and efficacy. Descriptive statistics were used to summarize the findings. A Wilcoxon signed-rank test was performed to assess change from baseline for immune cell counts and immunoglobulin levels. Only patients with values at baseline and for each follow-up visit were included, and multiplicity was not adjusted. Due to the many exploratory tests conducted, values of P < .05 were regarded as descriptive, not definitive. PFS was defined as time from first dose to documented progressive disease (PD) or death; DOR was defined as the time from the date of achieving the first response (partial response [PR] with lymphocytosis [PRL] or better) to the date of PD or death due to any cause, whichever occurred first. Values for PFS and DOR were estimated using Kaplan-Meier methodology48 ; PFS and DOR data were censored at the last clinical assessment for patients who discontinued treatment without documented PD or death. EFS was defined as time from first dose to documented PD, death, start of new anticancer therapy, or treatment discontinuation due to AE, whichever occurred first.
Results
Patients
Overall, 135 patients enrolled in this study between 30 January 2014 and 18 November 2015; 134 received ≥1 dose of acalabrutinib, and 130 patients had ≥1 postbaseline assessment. The median age was 66 years (range, 42-85 years); 20% were aged ≥75 years (Table 1). Patients received a median of 2 (range, 1-13) prior treatments, including chemoimmunotherapy (supplemental Table 1). Derived Rai staging at time of enrollment showed 49% had high-risk disease; 39% had bulky disease (lymph nodes ≥5 cm; 19% of which had nodes ≥10 cm). Sixty-nine percent of patients had cytopenia at baseline (ie, ANC, ≤1.5 × 109/L; hemoglobin, ≤11 g/dL; or platelet count, ≤100 × 109/L). β2-microglobulin >3.5 mg/L occurred in 75% of patients. Baseline genomic features included 73% of patients with IGHV unmutated disease, 23% with del(17)(p13.1), 18% with del(11)(q22.3), and 35% with complex karyotype.
Baseline demographic and clinical characteristics
| Characteristic | N = 134 |
|---|---|
| Median age (range), y | 66 (42-85) |
| Age, n (%), y | |
| ≥65 | 77 (57) |
| ≥75 | 27 (20) |
| Men, n (%) | 99 (74) |
| Diagnosis of CLL, n (%) | 132 (99) |
| ECOG performance status, n (%) | |
| 0 | 48 (36) |
| 1 | 82 (61) |
| 2 | 4 (3) |
| Bulky lymph nodes, n = 133, n (%), cm in diameter | |
| ≥5 | 52 (39) |
| ≥10 | 10 (8) |
| Rai risk classification (derived at screening), n (%)* | |
| Low | 0 |
| Intermediate | 38 (28) |
| High | 65 (49) |
| Missing | 31 (23) |
| Prior therapies, median (range) | 2 (1-13) |
| Cytopenia at baseline, n (%) | 92 (69) |
| ANC, ≤1500 µL | 28 (21) |
| Hemoglobin, ≤11.0 g/dL | 43 (32) |
| Platelet count, ≤100 000/µL | 65 (49) |
| Prognostic factor, n/N (%) | |
| Unmutated IGHV | 81/111 (73) |
| Chromosome 17p13.1 deletion | 27/116 (23) |
| Chromosome 11q22.3 deletion | 21/116 (18) |
| Complex karyotype, ≥3 abnormalities | 20/57 (35) |
| B2-microglobulin, >3.5 mg/L | 76/101 (75) |
| Characteristic | N = 134 |
|---|---|
| Median age (range), y | 66 (42-85) |
| Age, n (%), y | |
| ≥65 | 77 (57) |
| ≥75 | 27 (20) |
| Men, n (%) | 99 (74) |
| Diagnosis of CLL, n (%) | 132 (99) |
| ECOG performance status, n (%) | |
| 0 | 48 (36) |
| 1 | 82 (61) |
| 2 | 4 (3) |
| Bulky lymph nodes, n = 133, n (%), cm in diameter | |
| ≥5 | 52 (39) |
| ≥10 | 10 (8) |
| Rai risk classification (derived at screening), n (%)* | |
| Low | 0 |
| Intermediate | 38 (28) |
| High | 65 (49) |
| Missing | 31 (23) |
| Prior therapies, median (range) | 2 (1-13) |
| Cytopenia at baseline, n (%) | 92 (69) |
| ANC, ≤1500 µL | 28 (21) |
| Hemoglobin, ≤11.0 g/dL | 43 (32) |
| Platelet count, ≤100 000/µL | 65 (49) |
| Prognostic factor, n/N (%) | |
| Unmutated IGHV | 81/111 (73) |
| Chromosome 17p13.1 deletion | 27/116 (23) |
| Chromosome 11q22.3 deletion | 21/116 (18) |
| Complex karyotype, ≥3 abnormalities | 20/57 (35) |
| B2-microglobulin, >3.5 mg/L | 76/101 (75) |
ECOG, Eastern Cooperative Oncology Group.
Derived based on data collected at screening; patients with absolute lymphocyte count <5 × 109/L at screening were reported as missing.
Exposure and patient disposition
Patients initially received acalabrutinib at 100 (n = 9), 175 (n = 8), 250 (n = 7), and 400 mg once daily (n = 6) and 200 mg twice daily (n = 6) in the dose-escalation portion of the study, and 100 mg twice daily (n = 65) and 200 mg once daily (n = 33) in the phase 2 portion. Patients received acalabrutinib for a median of 41 months (range, 0.2-58 months); 26% of patients received acalabrutinib for >4 years. More than one-half of patients (n = 75; 56%) remained on treatment at data cutoff. For the 44% of patients (n = 59) who discontinued treatment, the reasons for discontinuation were PD (21%), AEs (11%), death (4.5%), physician decision (4.5%), withdrawal by patient (2.2%), and other (0.7%). Seventeen patients (13%) died; 10 deaths (7.5%) were due to AEs (detailed in “Safety”), and 7 (5%) were due to PD, including 2 patients (1.5%) with Richter transformation.
Pharmacodynamic studies
Median BTK occupancy at steady state (day 8) was 99% 4 hours postdose and 97% 12 hours postdose (at drug trough) for patients receiving acalabrutinib 100 mg twice daily (Figure 1); similarly, median occupancy at trough was 98% on day 28. In comparison, the 200-mg, once-daily regimen had a lower median occupancy at trough (24 hours postdose) of 94% and 95% at day 8 and day 28, respectively (Figure 1). Interpatient variability (coefficient of variation) on day 8 was 7% for twice-daily vs 14% for once-daily dosing.
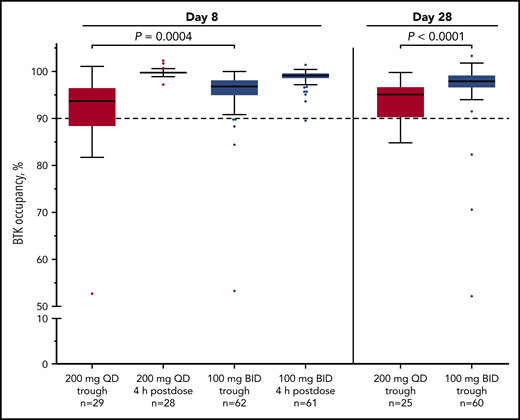
Twice-daily dosing results in near-complete target coverage over the dosing interval. BTK target occupancy enzyme-linked immunosorbent assay was performed on PBMC lysates using a biotin-tagged covalent analog of acalabrutinib as a molecular probe. Drug trough was 12 or 24 hours after dosing for the twice-daily (BID) and once-daily (QD) regimens, respectively. Box and whiskers show the median using the Tukey method for whisker range. Statistical significance was determined using an unpaired, parametric, 2-tailed Student t test.
Twice-daily dosing results in near-complete target coverage over the dosing interval. BTK target occupancy enzyme-linked immunosorbent assay was performed on PBMC lysates using a biotin-tagged covalent analog of acalabrutinib as a molecular probe. Drug trough was 12 or 24 hours after dosing for the twice-daily (BID) and once-daily (QD) regimens, respectively. Box and whiskers show the median using the Tukey method for whisker range. Statistical significance was determined using an unpaired, parametric, 2-tailed Student t test.
Patients with CLL can have chronic activation of T cells, inducing an exhausted state.49 Therefore, we evaluated whether acalabrutinib treatment improved T-cell response as measured by the production of the effector cytokine, interferon γ (IFNγ), in vitro (supplemental Figure 1). After 6 months of acalabrutinib therapy, the proportion of CD8+ T cells capable of producing IFNγ increased to a similar level as observed in T cells from healthy subjects (supplemental Figure 1A-B). This effect was particularly pronounced in patients with lower IFNγ production before therapy (supplemental Figure 1C; P < .01). Finally, we evaluated programmed death 1 expression on CD4+ T cells as a feature of T-cell exhaustion.50 The level of programmed death 1 expression on T cells decreased from 34.7% to 28.1% after 6 months of therapy (P = .03; supplemental Figure 2). Taken together, functional and phenotypic markers of T-cell exhaustion decreased during acalabrutinib treatment.
Serum immunoglobulin A (IgA) levels showed a trend toward normalization over time, whereas IgG and IgM levels did not (supplemental Figure 3). No clinically significant changes in T cell (CD4+, CD8+), natural killer cell, or monocyte numbers were observed (supplemental Figure 4).
Safety
All patients reported ≥1 AE. Supplemental Table 2 details AEs of all grades occurring in ≥10% of patients receiving acalabrutinib; generally, these were mild to moderate and were most commonly diarrhea (52%), headache (51%), upper respiratory tract infection (37%), and fatigue (34%). For patients who experienced diarrhea, headache, or fatigue, events were mostly self-limiting. Although 1 patient (1%) discontinued acalabrutinib due to diarrhea and 7 patients (5%) had dose holds for diarrhea, no changes to acalabrutinib administration were made due to fatigue; there was 1 dose hold for headache.
Grade ≥3 AEs occurred in 89 patients (66%), including 5 events that occurred in ≥5% of patients: neutropenia (14%), pneumonia (11%), hypertension (7%), anemia (7%), and diarrhea (5%; Figure 2A-B). Grade ≥3 infections occurred in 23% of patients (Figure 2C). Serious AEs occurring in ≥2 patients are listed in supplemental Table 3. Ten patients experienced grade 5 AEs: pneumonia (n = 5), Candida sepsis, congestive cardiac failure, metastatic lung adenocarcinoma, metastatic prostate cancer, and respiratory failure (n = 1 each). The overall frequency of grade ≥3 events of pneumonia, diarrhea, anemia, neutropenia, and thrombocytopenia decreased over time on acalabrutinib therapy (Figure 2D).
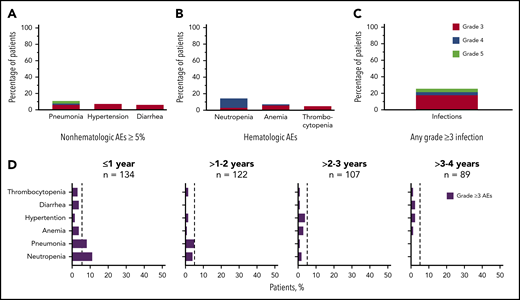
Frequency of grade ≥3 AEs. (A) All grade ≥3 nonhematologic AEs occurring in ≥5% of patients. (B) All grade ≥3 hematologic events. (C) All grade ≥3 infections. (D) Serial assessment of grade ≥3 AEs over time. The dashed line denotes 5% rate; time to onset is based on first occurrence of grade ≥3 event.
Frequency of grade ≥3 AEs. (A) All grade ≥3 nonhematologic AEs occurring in ≥5% of patients. (B) All grade ≥3 hematologic events. (C) All grade ≥3 infections. (D) Serial assessment of grade ≥3 AEs over time. The dashed line denotes 5% rate; time to onset is based on first occurrence of grade ≥3 event.
AEs of interest were assessed to better understand the safety profile of acalabrutinib. Major bleeding events (grade ≥3, serious AEs, and/or any central nervous system hemorrhage) occurred in 7 patients (5%); 4 of the events were grade 3 events (epistaxis, sublingual hematoma, gastric hemorrhage, and respiratory tract hemorrhage) and 3 were grade 2 (perirenal hematoma, rectal hemorrhage, and subarachnoid hemorrhage [due to fall from a bicycle]). Ten patients (7%) experienced atrial fibrillation (4 were grade ≥3; all others were grade ≤2) for a rate of 2.9 per 100 patient-years. Of the 10 patients who experienced atrial fibrillation, 7 had 1 or more risk factors for atrial fibrillation including age >65 years, hypertension, chronic obstructive pulmonary disease, arteriosclerotic coronary artery disease, or prior atrial fibrillation. The time to onset varied from 23 days on study (1 patient with prior atrial fibrillation who discontinued due to atrial fibrillation on study) to >1 to 3 years on study (all other patients).
Seventeen patients (13%) discontinued acalabrutinib due to AEs, including 4 events of pneumonia (all grade 5), and 2 events each of anemia (grade 1 and grade 2), neutropenia (both grade 3), and thrombocytopenia (both grade 4); all other events leading to discontinuation (atrial flutter, shortness of breath, metastatic lung adenocarcinoma, myelodysplastic syndromes, chronic myelogenous leukemia, congestive heart failure, diffuse abdominal pain, lymphedema, and diarrhea) occurred in 1 patient each. Dose reductions (taking a lower dose for ≥3 consecutive days) occurred in 36 patients (27%) and most commonly were due to patient error (n = 19 [14%]) and per-protocol changes to 100 mg twice daily (n = 13 [10%]). Six patients (4%) had a dose reduction due to an AE. Dose withholding, defined as missing dosing for ≥7 consecutive days, occurred in 84 patients (63%). The most common reason for dose withholding was per protocol for procedures (eg, planned surgery; n = 40 [30%]) followed by AEs (n = 33 [25%]). Fourteen patients (10%) missed dosing for ≥7 consecutive days due to patient error (ie, noncompliance unrelated to AEs).
Efficacy
The ORR was 94% (95% confidence interval [CI], 89% to 97%), including 6 patients (4%) with complete response (CR), 112 (84%) with a PR, and 8 (6%) with PRL (Table 2). The median DOR was not reached. The estimated 45-month DOR for PRL or better was 63% (95% CI, 52% to 72%). As with other BCR-signaling antagonists, acalabrutinib caused an initial rise in lymphocyte blood count; median time to initial PRL or better was 1.9 months (range, 1.4-43.7 months), whereas median time to initial response of PR or better was 4.7 months (range, 1.6-43.7 months). Response was also demonstrated as improvement vs baseline in cytopenias, with 37 of 43 patients (86%) who were anemic (hemoglobin, ≤11 g/dL), 27 of 28 (96%) patients who were neutropenic (ANC, ≤1.5 × 109/L), and 54 of 65 (83%) patients who were thrombocytopenic (platelets, ≤100 × 109/L) achieving normal values during therapy. Median time to recovery to normal hemoglobin, neutrophils, and platelets was 16.1 weeks (95% CI, 12-24), 1.1 weeks (95% CI, 1-2), and 16.1 weeks (95% CI, 6-32), respectively. Of the 28 patients who were neutropenic at baseline, 1 received granulocyte colony-stimulating factor on study. At baseline, 16 patients (12%) were red cell transfusion–dependent, and 5 patients (4%) were platelet transfusion–dependent. Of those evaluated for transfusion dependence on treatment (received ≥6 cycles of acalabrutinib),12 of 14 and 4 of 4 patients, respectively, became transfusion independent with acalabrutinib therapy; the other 2 patients discontinued treatment due to PD. Of the 11 patients (8%) with a history of autoimmune hemolytic anemia or immune-mediated thrombocytopenia before initiating acalabrutinib, 1 patient developed recurrence during therapy, which led to treatment discontinuation due to AE (anemia) after a PR.
Investigator-assessed best response to acalabrutinib
| N = 134 | ||
|---|---|---|
| n/n | % (95% CI*) or n (%) | |
| ORR: CR + PR+ PRL | 94 (89, 97) | |
| ORR: CR + PR | 88 (81, 93) | |
| Best response | ||
| CR | 6 (4) | |
| PR | 112 (84) | |
| PRL | 8 (6) | |
| Stable disease | 2 (1) | |
| PD | 2 (1) | |
| Unknown† | 4 (3) | |
| ORR by high-risk subgroup: CR + PR + PRL | ||
| del(17)(p13.1) | 25/27 | 93 (76, 99) |
| del(11)(q22.3) | 20/21 | 95 (76, 100) |
| Unmutated IGHV | 77/81 | 95 (88, 99) |
| Complex karyotype, ≥3 abnormalities | 18/20 | 90 (68, 99) |
| N = 134 | ||
|---|---|---|
| n/n | % (95% CI*) or n (%) | |
| ORR: CR + PR+ PRL | 94 (89, 97) | |
| ORR: CR + PR | 88 (81, 93) | |
| Best response | ||
| CR | 6 (4) | |
| PR | 112 (84) | |
| PRL | 8 (6) | |
| Stable disease | 2 (1) | |
| PD | 2 (1) | |
| Unknown† | 4 (3) | |
| ORR by high-risk subgroup: CR + PR + PRL | ||
| del(17)(p13.1) | 25/27 | 93 (76, 99) |
| del(11)(q22.3) | 20/21 | 95 (76, 100) |
| Unmutated IGHV | 77/81 | 95 (88, 99) |
| Complex karyotype, ≥3 abnormalities | 18/20 | 90 (68, 99) |
The 95% exact binomial CI.
Patients did not have on-treatment assessments.
For the entire population of treated patients, median PFS was not reached (Figure 3A); the estimated 45-month PFS was 62% (95% CI, 51% to 71%). Median EFS was not reached in the overall population (Figure 3C); the estimated 45-month EFS was 58% (95% CI, 48% to 67%). ORRs were 95% for patients with IGHV unmutated disease, 95% for patients with del(11)(q22.3), 93% for patients with del(17)(p13.1), and 90% for patients with complex karyotype (Table 2). Median PFS (95% CI) for patients with del(17)(p13.1) and complex karyotype was 36 months (21 to not estimable) and 33 months (17 to not estimable), respectively, and was not reached in patients with del(11)(q22.3) (Figure 3A). Median PFS was not reached for patients with unmutated or mutated IGHV (Figure 3B). Of the patients who relapsed (n = 36) with CLL (n = 32) or Richter syndrome (n = 4), 69% had unmutated IGHV, 33% had del(17)(p13.1), and 25% had complex karyotype compared with 73%, 23%, and 35% for the overall population, respectively.
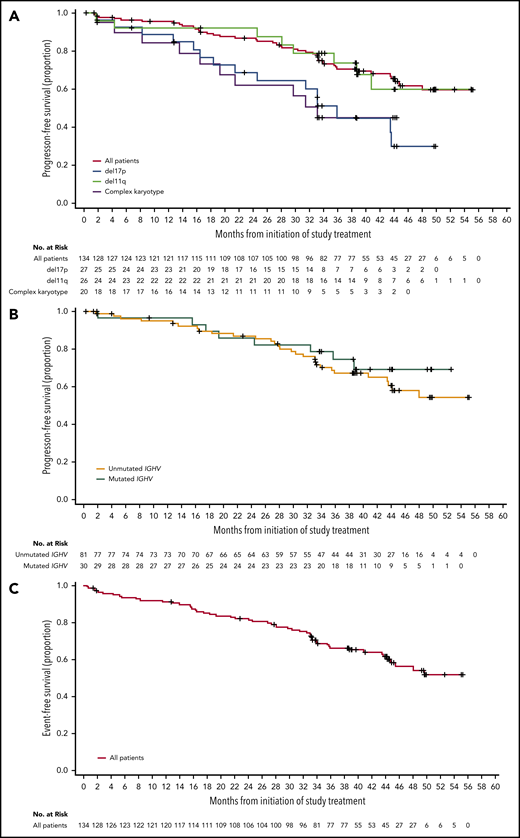
PFS and EFS. (A) Kaplan-Meier curves for PFS for all patients, and patients with del(17)(p13.1), del(11)(q22.3), or complex karyotype. (B) Kaplan-Meier curves for PFS based on IGHV mutational status. (C) Kaplan-Meier curve for EFS for all patients.
PFS and EFS. (A) Kaplan-Meier curves for PFS for all patients, and patients with del(17)(p13.1), del(11)(q22.3), or complex karyotype. (B) Kaplan-Meier curves for PFS based on IGHV mutational status. (C) Kaplan-Meier curve for EFS for all patients.
Genomic profiling
Nine patients had longitudinal PBMC samples from pretreatment baseline, during treatment, and at progression for whole-exome sequencing to assess acquired mechanisms of treatment resistance. Mutations of BTK were detected in 6 of 9 patients (67%) at time of progression; these mutations were not detectable at baseline (before acalabrutinib treatment), and were confirmed with droplet digital polymerase chain reaction (ddPCR) analysis of the C481 hotspot on BTK to ensure that exome analysis captured low-frequency mutations (Figure 4; supplemental Table 4). Of the 6 patients with detectable C581 mutations (5 C481S, 1 C481R), 4 had expansion to high-allele frequency of BTK mutation up to progression (up to 58%). In longitudinal analysis of patients who had a sample after 6 months of treatment (n = 3), the BTK mutation was not detectable. No PLCG2 gene mutations were detected using exome analysis in the 9 patients analyzed.

Analysis of BTK mutations in patients with R/R CLL who progressed during acalabrutinib treatment. (A) Summary of mutations detected in BTK across 2 different platforms. Text in parentheses indicates limits of mutation allele frequency (AF) detection. Each triangle represents a sample assessed at pretreatment baseline (left) or at progression (right); gray indicates wild-type BTK and green indicates BTK C481X mutation. (B) AF of BTK mutations in 6 patients with mutations at progression. Three time points are shown as a line plot for each patient and for each BTK mutation. The inset shows raw next-generation sequencing data from the integrated genome viewer of the BTK mutation at the progression time point. If 2 mutations coexisted in a single patient sample, separate reads are provided for each independent clone. Raw variant allele frequency (VAF) in Integrative Genomics Viewer (IGV) inset where filtered VAF plotted in line charts. PtID, patient identifier; WES, whole-exome sequencing.
Analysis of BTK mutations in patients with R/R CLL who progressed during acalabrutinib treatment. (A) Summary of mutations detected in BTK across 2 different platforms. Text in parentheses indicates limits of mutation allele frequency (AF) detection. Each triangle represents a sample assessed at pretreatment baseline (left) or at progression (right); gray indicates wild-type BTK and green indicates BTK C481X mutation. (B) AF of BTK mutations in 6 patients with mutations at progression. Three time points are shown as a line plot for each patient and for each BTK mutation. The inset shows raw next-generation sequencing data from the integrated genome viewer of the BTK mutation at the progression time point. If 2 mutations coexisted in a single patient sample, separate reads are provided for each independent clone. Raw variant allele frequency (VAF) in Integrative Genomics Viewer (IGV) inset where filtered VAF plotted in line charts. PtID, patient identifier; WES, whole-exome sequencing.
Discussion
We present updated results on an expanded cohort of patients from the initial phase 2 study and included long-term follow-up of the first cohort of patients with R/R CLL/SLL treated with acalabrutinib. These results confirm the early efficacy previously reported by doubling the number of patients initially reported. The response to acalabrutinib remains similar to this initial report and we now provide additional data on durability of response as well as long-term tolerability in most patients receiving this therapy. Notably, no late increase onset in infectious, metabolic, or hypertension was noted with acalabrutinib.
Acalabrutinib treatment produced a high ORR (90% to 95%) regardless of genomic characteristics (eg, IGHV mutation status, presence of del(11)(q22.3), del(17)(p13.1), or complex karyotype). Additionally, pretreatment cytopenias were largely improved and most patients gained transfusion independence with acalabrutinib treatment. After a median of 41 months of follow-up, the median DOR, EFS, and PFS had not been reached. PD occurred in 21% of patients, including 4 cases of Richter transformation (3%). Although the median PFS was not reached for the overall population, median PFS was 36 and 33 months for patients with del(17)(p13.1) and complex karyotype, respectively. A higher risk for PD for these genomic features has similarly been reported for the irreversible BTK inhibitor ibrutinib,7,51 highlighting an important unmet medical need.
Genomic profiling occurred in a small subgroup of patients with paired samples from baseline and progression (n = 9). Previously, 1 case of acquired BTK mutation was reported at the time of progression on acalabrutinib.45 Expansion of this analysis in the current study confirmed BTK mutation as the most frequent mechanism of acalabrutinib, with 67% of tested patients having a C481 amino acid substitution mutation at progression that was not detectable at baseline. No PLCG2 mutations were detected using whole-exome analysis, although deeper sequencing or ddPCR analysis may reveal low-frequency allele mutations. Further study of a larger number of acalabrutinib-treated patients will be required to ascertain whether a pattern of resistance different from ibrutinib exists.
Preclinical biochemical, in vitro, and in vivo studies demonstrate that acalabrutinib is a more selective BTK inhibitor than ibrutinib44 and, as such, may confer fewer AEs. In this study, AEs observed with acalabrutinib were generally mild to moderate, with a small subset of patients (11%) discontinuing therapy due to AEs. Events leading to discontinuation were most commonly infections and typically occurred early during treatment. As BTK inhibition requires chronic dosing until progression, the importance of long-term tolerability cannot be overstated, especially when treating a disease that occurs in the elderly, such as CLL. In addition, the favorable AE profile observed with acalabrutinib to date supports combination with other therapeutics for the treatment of CLL. Notably, acalabrutinib with follow-up beyond 4 years has shown a decreased frequency of AEs over time and no long-term safety issues to date. Comparison of acalabrutinib to other agents, such as ibrutinib, across different clinical trials is difficult and not preferred due to varied patient population, investigator awareness of toxicity, and differences in intervals between patient follow-up visits. An ongoing randomized phase 3 study of acalabrutinib vs ibrutinib in high-risk relapsed CLL (NCT02477696) will help differentiate the safety profiles of the 2 agents.
The selectivity of acalabrutinib and its short half-life offer the opportunity to apply more therapeutic inhibition of BTK, measured by the surrogate marker of BTK occupancy. During this course of the study, all patients were eventually switched to acalabrutinib 100 mg twice daily based on the efficacy, safety, and increased BTK occupancy of that dose and schedule. The twice-daily regimen yielded higher trough BTK occupancy with lower interpatient variability than the once-daily regimen. As with all covalent, irreversible inhibitors, the pharmacodynamic effect is largely determined by the de novo protein synthesis rate of the target protein. Reactivation of BTK signaling depends on newly synthesized BTK. Therefore, the twice-daily regimen ensures high target coverage, even when the BTK protein turnover rate is rapid. Less selective irreversible BTK inhibitors with a longer half-life, such as ibrutinib, are likely to be challenging to administer with twice-daily dosing. To date, there has been no such schedule of administration reported for ibrutinib.
Aside from a direct effect on CLL cells through BTK inhibition, acalabrutinib may also exert an effect on the tumor microenvironment. CLL is associated with defects in T-cell function, likely due to the persistent exposure to antigen by CLL cells.49 Thus, disease-related T-cell exhaustion may contribute to decreased antitumor immunity and increased infections.49,52 We observed abrogation of T-cell exhaustion features with acalabrutinib therapy, including expansion of CD8+ T cells capable of producing IFNγ to the levels measured in healthy cells. Because IFNγ is an important cytokine for driving T-cell effector functions, these results may represent a normalization of T-cell responsiveness. In addition, we previously reported that acalabrutinib suppressed plasma cytokine levels (eg, tumor necrosis factor α), which are commonly elevated in patients with CLL, thereby impacting tumor immunity.45 Similar to the report by Long et al,53 acalabrutinib was not associated with an overall expansion of T cells in this trial, which differs from their observations with ibrutinib. As ITK regulates T-cell migration, signaling, and activity, the lack of T-cell expansion likely reflects the lack of ITK inhibition with acalabrutinib; ITK inhibition is known to prevent activation-induced death. Changes to the humoral immune system were also observed in acalabrutinib-treated patients over time, with improvement in IgA levels, but not IgG or IgM levels. A similar effect on IgA levels over time was observed with ibrutinib in previously untreated CLL.54 This recovery of IgA levels, along with improvement in cellular immunity, may in part explain why infections with acalabrutinib and ibrutinib decrease over time. Taken together, the efficacy of BTK inhibition in CLL may be due to direct effects on CLL cells as well as indirect effects on the tumor microenvironment.55
In summary, this phase 2 study in patients with R/R CLL or SLL demonstrated favorable safety, response, and durability of response with acalabrutinib. An ongoing phase 3 study that compare acalabrutinib with standard first-line treatment (NCT02475681) will address the long-term contribution of this agent to the treatment armamentarium for CLL. In a phase 3 R/R CLL trial, acalabrutinib demonstrated superiority in prolonging PFS, with a more tolerable safety profile, compared with idelalisib plus rituximab or bendamustine plus rituximab.56 Several studies are also under way to assess the combination potential of acalabrutinib with other agents, including venetoclax and other targeted therapies for CLL, which ultimately may allow cessation of therapy at an appropriate treatment end point.
Acerta Pharma, a member of the AstraZeneca Group, is committed to data transparency and will consider data sharing requests on a case-by-case basis. Any requests for de-identified patient data can be submitted to Acerta Pharma 3 months post-publication and ending 5 years following article publication with the intent to achieve aims of the original proposal. In addition, Acerta Pharma will provide the study protocol, statistical analysis plan, informed consent form as well as post results on clinicaltrials.gov, as required.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the investigators and coordinators at each of the clinical sites, the patients who participated in this trial and their families, and the Acerta study team. Medical writing support (for figures, editing, and fact-checking) was provided by Tiffany Brake of Team 9 Science.
This work (including medical writing support for figures, editing, and fact-checking) was supported by Acerta Pharma, a member of the AstraZeneca Group. This work was also supported by grants from the National Institutes of Health, National Cancer Institute (R35 CA197734), the Leukemia & Lymphoma Society, the Kevin Sullivan Foundation, the D. Warren Brown Foundation, and the Four Winds Foundation. J.C.B. was supported by the Four Winds Foundation, the D. Warren Brown Foundation, Mr. and Mrs. Michael Thomas, the Sullivan CLL Research Foundation, and National Institutes of Health, National Cancer Institute grant R35 CA197734.
Authorship
Contribution: J.C.B. drafted and wrote the first version of this paper and revised subsequent versions; and all authors contributed to the design of the study and/or interpretation of data for this article, contributed to drafts of the article, and approved the final version to be published.
Conflict-of-interest disclosure: J.C.B. has received research funding from Acerta Pharma, Genentech, Janssen, and Pharmacyclics, and is a consultant (<$5000) for Acerta Pharma, Pharmacyclics, and Jazz Pharmaceuticals. W.G.W. has been a consultant for, and has received honoraria from, AbbVie, Celgene, Emergent, Genentech/Roche, Genzyme, Gilead, GlaxoSmithKline/Novartis, Sanofi, Merck, and Pharmacyclics; and has received research funding from Acerta Pharma, AbbVie, Emergent, Genentech, Gilead, GlaxoSmithKline/Novartis, Janssen, Juno, Karyopharm, Kite, and Pharmacyclics. A.S. has been a consultant for and has received honoraria from AbbVie, Gilead, GlaxoSmithKline, Janssen, Novartis, and Roche. S.D. has been a consultant for AbbVie, Gilead, GlaxoSmithKline, Janssen, MSD, and Roche; has received honoraria from AbbVie, Gilead, Janssen, and MSD; has received travel expenses from Gilead, Janssen, and Roche; is a member of speakers’ bureaus for Gilead and Janssen; and is on an advisory board for Servier. J.M.C. is an employee of Northwest Medical Specialties, PLLC. J.R.B. has been a consultant for AbbVie, Astellas Pharma, AstraZeneca, BeiGene, Loxo, Sunesis, Celgene, TG Therapeutics, Gilead, Verastem Pharmaceuticals, Janssen, Pfizer, Pharmacyclics, Redx, Roche/Genentech, and Sun BioPharma; has received research funding from Gilead, Verastem, Loxo, and Sun; and serves on a data safety monitoring board for Morphosys and Invacs. P.H. has been a consultant for and has received honoraria from AbbVie, Acerta Pharma, Alexion Pharmaceuticals, Gilead, and Janssen; and his institution has received research funding from AbbVie, Gilead, Janssen, GlaxoSmithKline, Pharmacyclics, and Roche. P.M. has been a consultant for Bayer, Genentech, Gilead, Janssen/Pharmacyclics, Novartis, and Verastem; and has received research funding from Celgene and Teva. F.T.A. has been a consultant for AbbVie, Janssen, Gilead, AstraZeneca, Sunesis, Genentech, Celgene, Blueprint Medicines, and Pharmacyclics; has received research funding from Innate Pharma and Pharmacyclics; and has served on speakers’ bureaus for AbbVie and AstraZeneca. D.M.S. has received research funding from Acerta, Gilead, Karyopharm, and the Lymphoma Research Foundation. P.G. has been a consultant for AbbVie, Acerta Pharma, AstraZeneca, Adaptive, ArQule, BeiGene, Dynamo, Gilead, Janssen, and Sunesis; and has received research funding from AbbVie, Gilead, Janssen, Novartis, and Sunesis. J.B. has served as a board or advisory committee member for AbbVie/Pharmacyclics, Gilead, and Janssen; and has received research funding from AbbVie/Pharmacyclics, Acerta Pharma, and Gilead. J.M.P. has been a consultant for Gilead and Pharmacyclics, and has equity ownership in and has received research funding from Actinium Pharmaceuticals, Inc. J.A.W. has been a consultant for Janssen, and received research funding from Acerta, AbbVie, Karyopharm, and Morphosys. P.P. is a current employee of and has equity ownership in Acerta Pharma; and has equity ownership in AstraZeneca. A.H., R.I., T.C., and M.G. are employees of, have equity ownership in, and hold related patents with Acerta Pharma. M.M.F. is an employee of, patent holder with, and equity owner of AstraZeneca. K.B. is an employee and equity owner of AstraZeneca. W.R. has equity ownership in, holds related patents with, and serves on the board of directors of Quogue BioVentures II LLC. M.H.W. is an employee of and has equity ownership in Acerta Pharma; and has equity ownership in AstraZeneca. S.O. has been a consultant for Amgen, Astellas, Celgene, GlaxoSmithKline, Janssen Oncology, Aptose Biosciences Inc, Vaniam Group LLC, AbbVie, and Alexion; has received research support from Kite Pharma, Regeneron, and Acerta Pharma; and has been a consultant for and received research support from Gilead, Pharmacyclics, TG Therapeutics, Pfizer, and Sunesis. R.R.F. has been a consultant for AbbVie, Acerta Pharma, AstraZeneca, BeiGene, Celgene, Gilead, TG Therapeutics, Verastem Pharmaceuticals, Janssen, Pfizer, Pharmacyclics, Roche/Genentech, Sunesis, and Loxo Pharmaceuticals; has served on the data safety monitoring boards of Incyte; has received research funding from Acerta Pharma, TG Therapeutics, Verastem, and Celgene; and has received speaker fees from Janssen.
Correspondence: John C. Byrd, The Ohio State University, 455B OSUCCC, 410 West 12th Ave, Columbus, OH 43210; e-mail: john.byrd@osumc.edu.
