

TET2 is one of the most commonly mutated genes in myeloid neoplasia. Somatic TET2 mutations (TET2MT) cause complete or partial loss of enzymatic activity. TET2 (along with TET1/3) are Fe2+ and αKG-dependent DNA-dioxygenases that catalyze the oxidation of 5mC→5hmC→5fC→5caC. Ultimately, 5hmC generated by TET2-dioxygenase passively prevents maintenance methylation due to DNA methyltransferase's inability to recognize 5hmC. Alternatively, demethylation may also be a result of base excision repair of fC and caC. TET2MT can serve as therapeutic targets because they are often initiating lesions and present in a large fraction of patients.
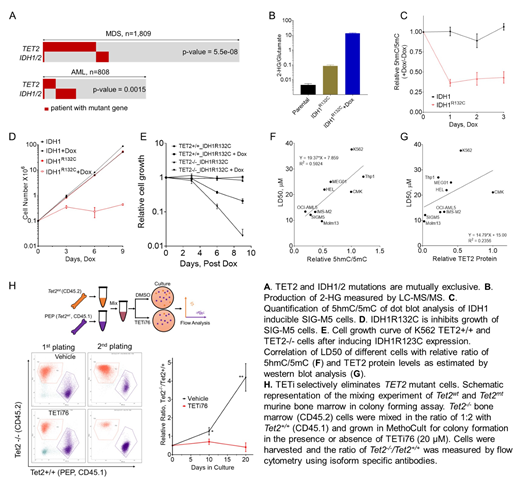
In this study, a comprehensive analysis of the configurations of TET2MT in myeloid neoplasia including MDS (n=1809) and AML (n=808), showed a remarkable exclusivity with 2-HG producing neomorphic IDH1/2MT (Fig.A). TET2 expression in 97 healthy and 909 MDS/MPN or AML patients from two independent studies showed that IDH1/2MT cases have significantly higher TET2 expression and were also mutually exclusive in cases with lower TET2 expression. Doxycycline inducible expression of IDH1MT led to profound growth inhibition of both a natural TET2MT cell line SIG-M5 and engineered TET2-/- K562, while the effect to parental K562 was mild (Fig.B-E). These observations suggest that mutual exclusivity of TET2MT and IDH1/2MT is due to synthetic lethality of TET2MT cell caused by 2-HG production, rather than redundancy of the consequences of IDH1/2MT and TET2MT.
In TET2MT cell 2-HG further inhibit the residual TET-activity (TET1/3) and may cause synthetic lethality to cells with affected TET2 function. SIG-M5 cells expresses significant amount of TET3 while negligible levels of TET1. The reliance on relative compensation through residual TET3 activity has been confirmed in cells by inducible TET3 knockdown. We hypothesized that transient suppression of the residual DNA dioxygenase activity with inhibitors may selectively eliminate TET2-deficient clones. The known TET inhibitors 2-HG, N-oxalylglycine (NOG) and dimethyl methyl fumarate (DMF) lack specificity, pharmacologic properties and potency. Based on the results of in silico docking simulations, we designed and synthesized 16 aKG derivatives. Among them, TETi76 showed best inhibition effect in both TET activity and cell growth of TET2 low expressing cell. TETi76 binds to the α-KG co-factor site of TET2 that principally involves H1801, H1381 and S1898. These amino acids are conserved in all three TET enzymes.
To test the in vitro efficacy and specificity of TETi, we used several human myeloid cell lines that harbor loss of function TET2 mutations or constitutively express low TET2 levels as well as bone marrow derived from Tet2+/+, Tet2+/- and Tet2-/- mice (Fig.F-G). Results showed that cells with low 5hmC level were more sensitive to TETi76 treatment. Specificity of TETi76 was further confirmed by RNAseq analyses of TETi76 treated K562, TET2-/- K562 and parental control cells. Moreover, TETi treatment did not appear to affect the function of α-KG-dependent histone dioxygenases.
Mechanistically, treatment of SIG-M5 cells with TETi76 induced early and late stages of apoptotic cell death, a finding further confirmed by PARP1 and caspase-3 cleavage. RNAseq analyses of SIGM5 cells after treatment with TETi demonstrated a significant down-regulation of genes involved in transcription and peptide elongation, consistent with the consequences of TET inhibition. Interestingly, we also observed significant up-modulation of oxidative stress response pathway genes consistent with the inhibition of dioxygenases. In particular, TETi76 treatment induces 8-fold increase of oxidative stress sensor NQO1 a NRF2 target gene.
To further probe the effects of TETi76 on TET2 deficient cells, Tet2MT/Tet2WT BM cells were co-cultured at fixed ratios to mimic the evolving Tet2MT clones. TETi76 effectively eliminated otherwise dominating Tet2MT cells (Fig.H). To determine the in vivo effects of TETi e.g., on elimination of Tet2MT clones, we performed bone marrow competitive reconstitution assays in PEP mice. TETi treatment selectively restricted the proliferative advantage of Tet2MT HSC compared to vehicle control where, as expected, TET2 mutant clones took over the WT cells. In clinical applications, TET inhibitors may constitute a new class of agents to be used in a targeted fashion in TET2 mutant neoplasia.
Meggendorfer:MLL Munich Leukemia Laboratory: Employment. Abazeed:Bayer AG: Honoraria, Other: Travel Support, Research Funding; Siemens: Research Funding. Sekeres:Millenium: Membership on an entity's Board of Directors or advisory committees; Syros: Membership on an entity's Board of Directors or advisory committees; Celgene: Membership on an entity's Board of Directors or advisory committees. Haferlach:MLL Munich Leukemia Laboratory: Employment, Equity Ownership. Maciejewski:Novartis: Consultancy; Alexion: Consultancy.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal