

Deregulation of transcription is a hallmark of acute myeloid leukemia (AML) that drives oncogenic expression programs and presents opportunities for therapeutic targeting. We hypothesized that by integrating the active enhancer chromatin landscapes with pan-cancer enhancer and genetic dependency mapping, we would be able to identify targetable tumor-specific oncogenic enhancer regulation and gain mechanistic insights into the underlying basis of the regulation.
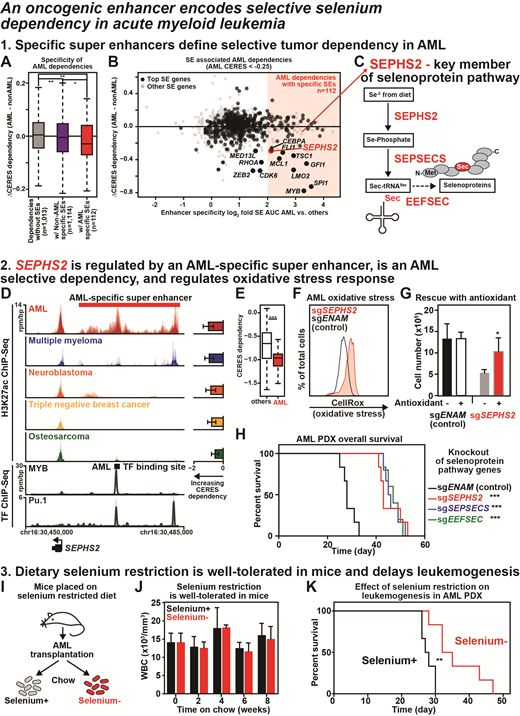
Using this approach, we find that tumor-specific super enhancers statistically demarcate tumor-specific dependency (Fig. A). In addition to several well-known AML oncogenes (e.g. MYB, CDK6, FLI1…), we find SEPHS2 to have highly AML-specific enhancer regulation and genetic dependency (Fig. B). SEPHS2 is a key component of the selenoprotein production pathway and is required for the selenocysteine incorporation during protein translation (Fig. C).
Across AML cell lines and primary patient samples, we observe a large AML-specific super enhancer at the SEPHS2 locus (Fig. D) that correlates with SEPHS2 genetic dependency (Fig. E) and includes a prominent binding site for AML transcription factors (Fig. D). We confirmed that the oncogenic transcription factor MYB strongly regulates SEPHS2 expression and that SEPHS2 expression correlates with poor prognosis in AML (not shown in this figure). Selenoproteins play an important role in mediating oxidative stress. We find that SEPHS2 knockout increases oxidative stress and that antioxidant treatment rescues viability effects of SEPHS2 knockout (Fig. F, G). Across murine and human in vivo AML models, genetic perturbation of selenoprotein production pathway genes strongly delays leukemogenesis (one example in Fig. H). Other cell lines (both cancerous and non-cancerous) are minimally affected by SEPHS2 knockout, confirming specificity in AML (Fig. E).
As a druggable enzyme SEPHS2 merits strong consideration as a therapeutic target. In the interim, we hypothesized that selenium dietary restriction (Fig. I) may be able to phenocopy selenoprotein pathway inhibition with a lower regulatory burden. We show that mice tolerate selenium-depleted chow with no observable effects on body weight or hematopoiesis (one example in Fig. J). Confirming our hypothesis, AML transplanted into these mice exhibit a strong delay in leukemogenesis (Fig. K).
Throughout the cancer biology field, there is broad interest in understanding how best to leverage pan-cancer genetic dependency data. We find that the integration of enhancer data adds another layer of specificity and helps provide mechanistic insight into the underlying basis of oncogenic deregulation and dependency. Our identification of AML-specific enhancer regulation of selenoprotein production - which has been minimally studied in this disease - validates our unbiased approach and points to selenoprotein production as a deregulated and therapeutically-actionable metabolic axis in AML.
Lin:Syros Pharmaceuticals: Equity Ownership, Patents & Royalties.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal