

Mutations in the RNA splicing factor SF3B1 are common in MDS and other myeloid malignancies. SF3B1 mutations promote expression of mRNAs that use an aberrant, intron proximal 3' splice site (ss). Despite the consistency of this finding, linking aberrant splicing changes to disease pathogenesis has been a challenge. Here we identify aberrant splicing and downregulated expression of BRD9, a member of the recently described ATP-dependent non-canonical BAF (ncBAF) chromatin remodeling complex, across SF3B1 mutant leukemias. In so doing, we identify a novel role for altered ncBAF function in hematopoiesis and MDS.
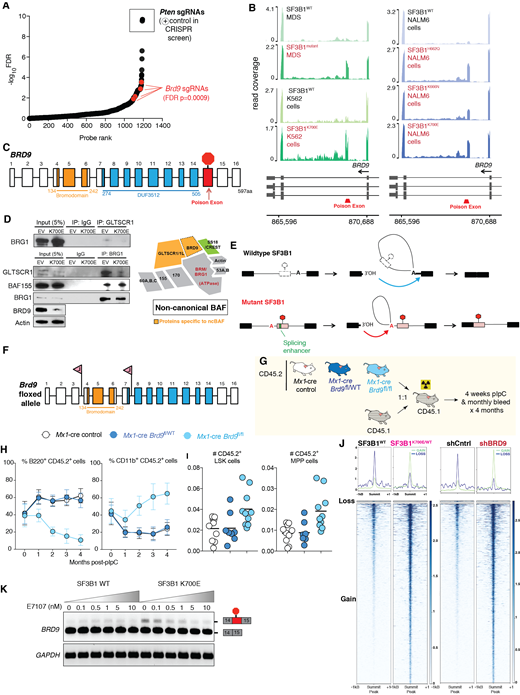
To systematically identify functionally important aberrant splicing events created by mutant SF3B1, we integrated differential splicing events in SF3B1 mutant versus wild-type MDS with a positive enrichment CRISPR screen mimicking splicing changes induced by mutant SF3B1 that promote NMD (non-sense mediated mRNA decay). We tested whether loss of any gene functionally inactivated by SF3B1 mutations promoted transformation of Ba/F3 and 32D cells. This identified a specific NMD-inducing aberrant splicing event in BRD9 which promoted cytokine independence (Fig. A) and exhibited striking aberrant splicing across CLL and MDS and across all mutational hotspots in SF3B1 (Fig. B). SF3B1 mutations cause exonization of a normally intronic sequence in BRD9, resulting in inclusion of a poison exon that interrupts BRD9's reading frame (Fig. C) and reduced BRD9 mRNA and protein expression through NMD (Fig. D). We confirmed that mutant SF3B1 suppressed full-length BRD9 levels without generating truncated BRD9 protein. Loss of BRD9 impaired ncBAF complex formation as indicated by abolished interaction between the ncBAF specific component GLTSCR1 and the ATPase subunit BRG1 upon chemical or spliceosomal BRD9 ablation (Fig. D).
Given that prior work has linked mutant SF3B1 to use of aberrant 3' ss, we sought to understand the molecular basis for aberrant exon inclusion in BRD9 by mutant SF3B1. Lariat sequencing of SF3B1 mutant versus WT K562 cells and BRD9 minigene analyses identified use of a deep intronic branchpoint adenosine by mutant SF3B1 to promote BRD9 poison exon inclusion (Fig. E).
The data above suggest a role for BRD9 downregulation in SF3B1 mutant leukemia. While prior work indicated that BRD9 is required in MLL-rearranged AML (Hohmman et al. Nature Chemical Biology 2016), the role of BRD9 in normal hematopoiesis or other subtypes of myeloid neoplasms has not been evaluated. Genetic downregulation of BRD9 in normal human hematopoietic progenitors from cord blood promoted myelopoiesis while impairing megakaryopoiesis. Interestingly and unexpectedly, BRD9 loss in CD34+ cells promoted terminal erythroid differentiation in vitro. To further evaluate BRD9's role in hematopoiesis in vivo, we also generated mice with inducible knockout of the bromodomain of BRD9 (required for BRD9 function) and generation of a frameshift transcript resulting in reduced Brd9 expression (Fig. F). Loss of Brd9 resulted in macrocytosis with bone marrow erythroid dysplasia in a dosage-dependent manner, along with impaired lymphopoiesis and myeloid skewing. Moreover, competitive transplantation of hematopoietic precursors from these mice revealed that ablation of Brd9 function impaired lymphoid reconstitution while promoting advantage of myeloid cells and hematopoietic precursors (Fig. G-I).
In myeloid leukemia cells, introduction of SF3B1K700E or downregulation of BRD9 resulted in increased chromatin accessibility at promoters with a significant overlap in commonly upregulated genes. This finding suggests shared epigenetic effects of SF3B1K700E mutations and BRD9 loss (Fig. J).
These data identify aberrant splicing of BRD9 across the spectrum of SF3B1 mutant cancers and identify a novel role for downregulation of ncBAF function in MDS pathogenesis. Consistent with human genetic data, genetic ablation of BRD9 function in mouse and human hematopoietic cells resulted in myeloid skewing and dyserythropoiesis. These data suggest that targeted correction of aberrant BRD9 splicing might serve as a novel therapeutic approach for SF3B1-mutant leukemias. Of note, treatment with drugs impairing the binding of mutant SF3B1 to RNA resulted in a dose-dependent rescue of aberrant BRD9 splicing in vitro (Fig. K) and in treatment of an SF3B1 mutant AML patient-derived xenograft in vivo.
Kadoch:Foghorn Therapeutics: Consultancy, Equity Ownership, Membership on an entity's Board of Directors or advisory committees.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal