

There is a need to understand and counter mechanisms-of-resistance to decitabine (Dec) and 5-azacytidine (5Aza), the only agents approved to treat all subtypes of myelodysplastic syndromes (MDS). Both Dec and 5Aza are pro-drugs, and here, in vitro, in vivo and patient data show that while processing the pro-drugs, pyrimidine metabolism is itself altered, dampening pro-drug conversion and causing resistance. Anticipation of these adaptive responses of the pyrimidine metabolism network enables their exploitation instead.
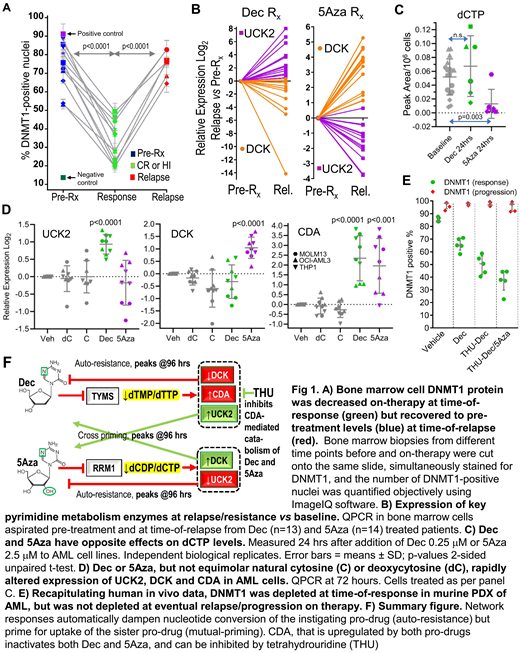
Dec and 5Aza are processed by pyrimidine metabolism into a nucleotide analog that depletes DNA methyltransferase 1 (DNMT1). We found that DNMT1 protein, although substantially depleted (~50%) in patients' bone marrows at time-of-response, rebounded at time-of-relapse (Fig 1A). The pyrimidine metabolism enzymes DCK and UCK2 rate-limit cellular uptakes of Dec and 5Aza, respectively. At relapse on Dec (n=13), DCK was halved vs patients' baseline bone marrows (Fig 1B). At relapse on 5Aza (n=14), it was UCK2 that was halved (Fig 1B). Simultaneous with DCK or UCK2 suppression, however, the other enzyme (UCK2 or DCK) was upregulated up to 40-fold (Fig 1B). Seesaw DCK and UCK2 expression was observed also in DCK knock-out (KO) and UCK2-KO AML cells (HAP1): DCK-KO cells upregulated UCK2, resisted Dec, but were more sensitive to 5Aza. UCK2-KO cells upregulated DCK, resisted 5Aza, but were more sensitive to Dec (concentrations for 50% growth inhibition [GI50] Dec: HAP1 wildtype 3mM, DCK-KO 12mM, UCK2-KO 0.1mM; 5Aza: wildtype 4mM, DCK-KO 2mM, UCK2-KO 15mM). These compensatory upregulations of UCK2 or DCK were adaptive to DCK- or UCK2-KO-induced decreases in dCTP or dTTP respectively. We thus examined if Dec or 5Aza also perturbed dCTP/dTTP amounts to trigger adaptive metabolic shifts. Dec decreased dTTP and increased dCTP, while 5Aza decreased dCTP, 24 hours after their addition to AML cells (THP1, OCI-AML3, MOLM13, K562)(Fig 1C). A single dose of Dec or 5Aza was also sufficient to upregulate UCK2 or DCK >2-fold, and the catabolic enzyme CDA >4-fold, within 72 hours (Fig 1D). Protein levels tracked the mRNA changes. Exponentially proliferating Dec or 5Aza-resisting AML cells emerged within 35 days in presence of pro-drug (5 AML cell lines) and displayed the same metabolic reconfigurations seen acutely. We then used patient-derived xenotransplant (PDX) models of chemorefractory AML to identify methods to counter this automatic metabolic adaptation or 'auto-resistance'. The Dec and 5Aza doses used (subcutaneous Dec 0.2 mpk or 5Aza 2 mpk) were chosen to deplete DNMT1 from marrow without cytotoxicity, demonstrated by flow cytometry for DNMT1 and gH2AX. We found (i) Dec timed to avoid DCK troughs (D1 & 2 each week) was superior to Dec timed to coincide with DCK troughs (D1 & 4 each week)(median survival 74 vs 62 days [vehicle 40 days], Log-rank p=0.006); (ii) Adding a CDA inhibitor (intraperitoneal tetrahydrouridine 4-10 mpk) to halved doses of Dec or 5Aza (0.1 mpk, 1 mpk respectively, to avoid cytotoxicity) was superior to no CDA inhibitor + Dec 0.2 mpk or 5Aza 2 mpk (median survival 180 vs 115 days [vehicle 50 days], Log-rank p=0.004), and (iii) Alternating Dec and 5Aza timed to peaks of mutual priming (e.g., D1 & 4 each week) was superior to Dec alone or 5Aza alone, or Dec/5Aza alternating every month or Dec/5Aza given together - the regimen incorporating these lessons extended survival by several months vs vehicle (median survival 223 vs 50 days, Log-rank p=0.003). Eventual AML relapse/progression on this therapy was again caused by AML cells in which DNMT1 was not depleted (Fig 1E), because of even larger increases in CDA and upregulation of de novo pyrimidine synthesis. We are thus evaluating higher CDA-inhibitor doses (40 mpk) and incorporation of non-cytotoxic doses of a de novo pyrimidine synthesis inhibitor.
In sum, the pyrimidine metabolism network responds automatically to Dec or 5Aza perturbation with adaptive shifts that decrease pro-drug conversion into the DNMT1-depleting nucleotide. These metabolic shifts can be anticipated (Fig 1F) and exploited, using simple, clinically viable treatment modifications.
Maciejewski:Novartis: Consultancy; Alexion: Consultancy. Saunthararajah:EpiDestiny: Consultancy, Equity Ownership, Patents & Royalties; Novo Nordisk: Consultancy.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal