

Background:
Originally described as a monogenic hemoglobin disorder resulting in increased red blood cell (RBC) stiffness leading to vaso-occlusion, sickle cell disease (SCD) is now known to be a vasculopathic disease with some semblance to cardiovascular disease in which the endothelium is inflamed. While adhesive RBC-endothelial interactions, inflammatory cytokines, and hemolysis all contribute to SCD vasculopathy, whether the increased stiffness of sickle RBCs directly contributes to endothelial inflammation is unknown.
Endothelial cells are now known to mechanotransduce shear forces into biological signals. Pathological alteration of such forces leads to proinflammatory endothelial cell signaling including upregulation of VCAM-1 and E-selectin, which contribute to atherosclerotic plaques leading to myocardial infarction and stroke (Abe, ATVB, 2014). In addition, under normal homeostatic conditions, RBCs do not come into contact with the endothelium due to a cell-free layer created by the Fåhræus-Lindqvist effect. Studies including our own have shown in silico that increasing RBC stiffness diminishes or eliminates the cell-free layer, allowing stiff RBCs to contact the vessel wall (Kumar, Phys Rev E, 2011). This is particularly pertinent in SCD, as all patients have a small population (1-10%) of sickle RBCs that are permanently stiff and misshapen. We therefore hypothesize that purely physical interactions - akin to "scratches" or collisions - between endothelial cells and stiff SCD RBCs breaking through the cell-free layer are sufficient to cause endothelial inflammation in the absence of adhesion or vaso-occlusion (Fig. 1A).
Methods:
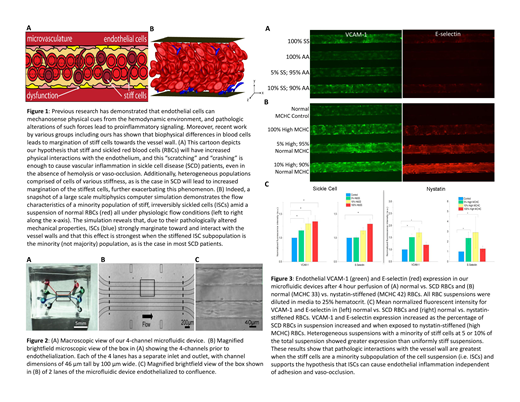
We performed computational direct numerical simulations using the boundary integral method for a binary suspension of flexible biconcave discs and stiff curved prolate spheres modeling healthy RBCs and ISCs, respectively. Experimentally, we leveraged our microfluidic microvasculature models of human umbilical vein endothelial cells cultured throughout each microchannel (Fig. 2). RBCs from SCD patients were "spiked" into normal RBC suspensions to comprise 5 and 10% of the overall population (a representation of ISCs in vivo), suspended in media to 25% hematocrit mimicking conditions seen in SCD patients, and perfused into the microfluidics for 4 hours. Samples of 100% normal RBCs or SCD RBCs were run in parallel. To isolate the stiffness effects of sickle RBCs without confounding hemolytic and adhesive effects, parallel experiments were conducted using nystatin-treated normal RBCs to create artificially stiffened RBC subpopulations, defined by elevated mean corpuscular hemoglobin concentrations (MCHCs), at the same proportion of the overall RBC population (0, 5, 10 and 100%). The endothelialized models were then fixed, permeabilized, and immunostained with antibodies against VCAM-1 and E-selectin. Mean fluorescence intensity was measured to quantify endothelial inflammation.
Results:
In silico, we observed that ISCs strongly marginate towards the vessel walls due to their stiffness and "pointy" shape, and heterogeneous suspensions with small fractions of stiff, pointy cells (5 and 10%) caused the highest degree of margination (Fig. 1B). Experimentally, endothelium exposed to 5, 10, and 100% SCD RBCs exhibited increased VCAM-1 and E-selectin expression over normal RBCs, and the degree of expression increased with higher percentages of SCD RBCs. While endothelial cells exposed to nystatin-stiffened RBCs also showed increased VCAM-1 and E-selectin expression, those exposed to a lower percentage of stiff cells (5 and 10%) exhibited higher expression than the homogenously stiff (100%) condition (Fig. 3), which is consistent with our computer simulations.
Conclusions:
Here we demonstrate that purely non-adhesive, physical interactions between endothelial cells and SCD RBCs are sufficient to cause endothelial inflammation. Furthermore, heterogeneous RBC populations, comprised of a small minority of stiff cells, cause more inflammation than uniformly stiff RBCs. Studies elucidating the underlying mechanisms, using different endothelial cell types, and analyzing the effect of vessel curvature are ongoing. Our results introduce a new paradigm for understanding SCD pathophysiology and may help explain how chronic diffuse vasculopathy develops, which could lead to more biophysically-based therapeutic strategies.
Carden:GBT: Honoraria; NIH: Research Funding. Mannino:Sanguina, LLC: Employment, Equity Ownership. Lam:Sanguina, LLC: Equity Ownership.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal