

Multiple myeloma (MM) is a hematological malignancy characterized by an abnormal accumulation of clonal plasma cells in the bone marrow. MM heterogeneity is associated to the presence of different genomic and transcriptomic profiles that have a clear impact on the prognosis of the disease. Metabolism has been deeply studied in cancer research, unveiling several vulnerabilities in different tumors. However, information regarding the role of metabolism in the pathogenesis of MM has not been explored in detail. Previous studies from our group using Systems Biology approach, explained the essentiality of metabolic gene RRM1 in several MM cell lines. The aim of our current study was to identify metabolic vulnerabilities in MM based on the application of a system biology approach focus on metabolic networks and trascriptomic data from MM patients. Our hypothesis being that changes in the metabolic landscape of MM could be exploited to uncover novel targets for prognosis and treatment in MM patients.
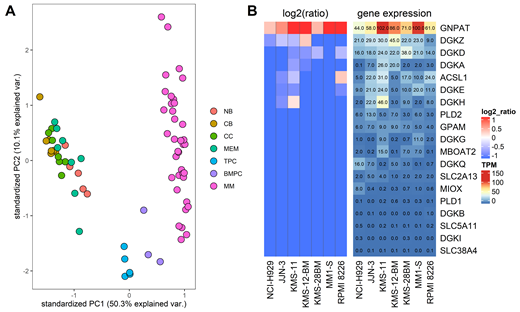
We have analyzed the strand specific RNA-seq data from 35 samples from different subpopulations of B cells (Naïve, Centroblast, Centrocyte, Memory, Tonsilar and Bone Marrow Plasma cells (PCs)) and PCs of 37 MM patient samples. Using only the expression of 3287 metabolic genes included in Recon3D, the latest human metabolic reconstruction, we identified metabolic transcription patterns that clearly differentiate the different B cells from MM plasma cells (Figure 1A). MM samples were more similar to the normal PCs than to other B cells, which suggest that they maintain part of the metabolic pattern of the PCs, but in turn showed significant differences in the expression of metabolic genes. Interestingly, differential expression analysis of metabolic genes in MM PCs revealed a decrease in the expression of genes involved in mitochondrial activity and an increase in those that participate in metabolic proliferation, indicating that these alterations could probably play an important role in the development of this tumor.
Using our novel systems biology approach, based on Recon3D and transcriptomic profiles, we predicted essential genes and synthetic lethals (involving two or more genes) that were specific for MM patients and not for the rest of the B cell subpopulations. Our approach makes use of the concept of genetic Minimal Cut Sets, previously introduced by our group in cancer research, which defines subsets of genes that if knocked out at the same time, induce a blockage of cellular proliferation. A metabolic vulnerability is found when only one gene is highly expressed in one of these gMCSs. We also analyzed the essentiality of these genes in more than 500 MM patients samples included in CoMMpass project and MM cell lines analyzed in Cancer Cell Line encyclopedia (CCLE). Using this computational strategy, we detected 8 essential genes involved in 42 gMCS specific for MM patients. From those candidates, GNPAT was our most promising target gene, as it was predicted to be essential for more than 40% of MM patients in our group, approximately 10% of patients of CoMMpass and in the majority of MM cell lines (Figure 1B). Validation of GNPAT expression and other 18 genes involved in the GNPAT gMCS was carried out by RT-qPCR showing similar results that were obtained with our RNA-seq data. Interestingly, the expression of the partner genes included in the GNPAT gMCS, such as DGK gene family, could also be indicators of the effectiveness of knocking-out, where their high expression is an indicator of resistance and their low expression an indicator of sensitivity.
In conclusion, our findings suggest that our systems biology computational approach, driven by RNA-seq data, identifies metabolic vulnerabilities (defined as essential genes or synthetic lethal genes) providing pairs of new targets (essential gene) and their associated biomarkers (gMCS) in patients with MM.
Figure 1: Metabolic genes expression analysis in human humoral immune response and MM patient samples. A) Standardized PCA result using the expression of metabolic genes included in Recon3D, in distinct subpopulations of B cells and MM samples. B) GNPAT gMCS showing the expression (in TPM) of GNPAT and associated 18 genes in MM cell lines.
Paiva:Amgen, Bristol-Myers Squibb, Celgene, Janssen, Merck, Novartis, Roche, and Sanofi; unrestricted grants from Celgene, EngMab, Sanofi, and Takeda; and consultancy for Celgene, Janssen, and Sanofi: Consultancy, Honoraria, Research Funding, Speakers Bureau. Melnick:Epizyme: Consultancy; KDAc Therapeutics: Membership on an entity's Board of Directors or advisory committees; Constellation Pharmaceuticals: Consultancy; Janssenn: Research Funding. San-Miguel:Amgen, Bristol-Myers Squibb, Celgene, Janssen, MSD, Novartis, Roche, Sanofi, and Takeda: Consultancy, Honoraria.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal