

Introduction: Additional Sex Combs-Like 1 (ASXL1) is a chromatin modifier frequently affected by truncating mutations in myeloid malignancies. These mutations are associated with poor survival outcomes and increased rates of acute leukemic transformation. In chronic myelomonocytic leukemia (CMML), ASXL1 mutations are thought to affect transcriptional activity mainly by modifying histone marks, however additional epigenomic mechanisms have not been fully explored. We interrogated the epigenome of patients with ASXL1-mutant (MT) and -wildtype (WT) CMML using a multiomics approach to define cis-regulatory elements (CREs) such as distal enhancers (DEs).
Methods: Bone marrow mononuclear cells from patients with CMML were subjected to targeted NGS of DNA, whole transcriptome shotgun sequencing (RNA-seq), immunoprecipitation (IP) of DNA (hydroxy-)methyl residues (DIP-seq), IP of the histone modifications H3K4me1, H3K4me3, and H3K27me3 (ChIP-seq), and DNA transposase accessibility studies (ATAC-seq). After quality control all samples were sequenced on an Illumina HiSeq 4000 before further processing and data analysis. Global assessments of DNA (hydroxy-)methylation, DNA accessibility, and histone modifications between ASXL1 MT and WT CMML were performed. The samples in the two groups were treated as biological replicates and subjected to a consensus peak calling strategy requiring an overlap of at least 30% between samples and an adjusted p-value < 5x10-5 for a signal peak to be considered statistically significant. Differential gene expression was estimated to define the up-regulated genes in ASXL1 MT CMML. Potential CREs were defined as sites with statistically significant signal peaks overlapping in at least two of the three epigenomic marks: H3K4me1, 5hmC, and ATAC. Potential DEs were defined as CREs in non-coding regions outside promoter regions (defined as transcription start site ±3kb) that were annotated in GeneHancer. Annotated DEs only present in ASXL1 MT but not WT CMML (specific DEs) were intersected with the list of up-regulated genes and the ReMap atlas.
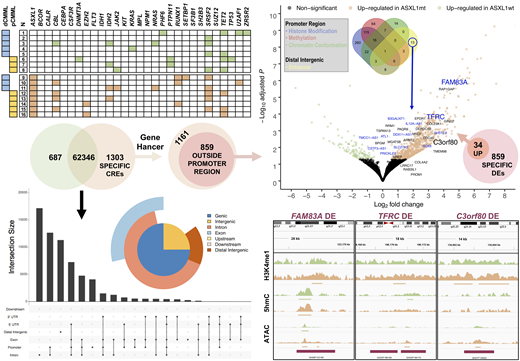
Results: Sixteen WHO-defined CMML patients were included, median age 69 years (48 - 77), 63% male; of which 8 patients (50%, all truncating frame shift mutations) were ASXL1 MT and 8 (50%) WT. The burden and spectrum of co-mutations was similar between ASXL1 WT and MT CMML (21 versus 23 per group; p = 0.684; heatmap). There was a predominant up-regulation of gene expression in ASXL1 MT CMML: 707 genes up- and 124 down-regulated (volcano plot, FDR < 0.050 for all genes). There were 64336 potential CREs, the vast majority (97%) being present in both ASXL1 MT and WT CMML (left Venn diagram). These CREs were most commonly located in introns, promoter regions, and distal non-coding regions (bar graph and pie chart). There were 1303 CREs unique to ASXL1 MT CMML (specific DEs), 1161 (90%) of which were annotated in GeneHancer (left Euler diagram). Of these 1161 annotated specific DEs 859 (74%) were located outside promoter regions and 34 (4%) of them were known to be associated with genes up-regulated in ASXL1 MT CMML (Euler diagrams). These specific DEs were characterized by an increase in H3K4me1 occupancy and DNA accessibility (average signal tracks, purple bars indicating annotated DEs, thin bars below peaks indicating statistical significance). We previously observed epigenomic modification of promoter regions in 519 of the 707 up-regulated genes (73%) facilitating transcriptional activity in ASXL1 MT CMML. For 13 of the up-regulated genes (right Venn diagram, blue genes in volcano plot) the specific DEs were the sole identified mechanism, while for the other 21 genes there were additional mechanisms noted in the promoter region. The top five transcription factor candidates binding the 34 specific DEs included JMJD1C, MYC, KDM5B, RCOR1, and HDAC2 (-log10(E) > 40 for all candidates).
Conclusions: Using a multiomics approach based on H3K4me1, 5hmC, and ATAC data we identified potential CREs in ASXL1 MT CMML and characterized potential DEs using publicly available annotation data. Specific DEs were associated with up-regulated genes serving as a possible explanation for the observed transcriptional activity, shedding further light on the adverse prognostic impact associated with ASXL1 mutations.
Patnaik:Stem Line Pharmaceuticals.: Membership on an entity's Board of Directors or advisory committees.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal