

MPN-BP originates from a leukemic stem cell (LSC) that is capable of recreating and serial passaging the leukemia in NSG mice (Wang Blood 2018). The genomic architecture of these LSCs has not been well characterized. We therefore performed mutational profiling using capture-based next generation sequencing of primary MPN-BP patient samples and xenografts following their transplantation into NSG mice.
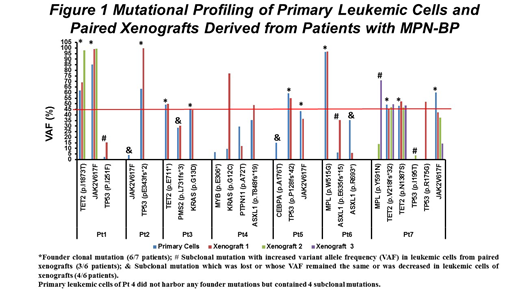
All 7 patients with MPN-BP studied had 2-6 known oncogenic gene mutations. T cell-depleted mononuclear cells (MNC, 1-10×106) containing 50-3850 leukemia initiating cells based upon limiting dilution analysis were transplanted into 1-3 NSG mice per patient sample. 1-7 months after transplantation, the mice were sacrificed. 23.3±5.5% leukemic cell chimerism was detected in each mouse. Leukemic cells from xenografts were then selected and sequenced. As shown in Figure 1, primary MNCs from 6 of 7 patients contained 1-3 mutations involving TET2, JAK2V617F, TP53, MPL, KRAS with a variant allele frequency (VAF) ≥45% marking founder clones. Leukemic cells in primary xenografts possessed each of their founder mutations, suggesting clonality and that MPN-BP originates within the LSC compartment. Additionally 1-2 gene mutations with 0.1-9.6% VAF (subclonal mutations) were present in primary samples from 3 of these 6 patients (Pts 1, 6, 7) but their VAF (15.5-71.1%) in xenografts surpassed that present in primary cells. Moreover, a distinct oncogenic mutation in TP53(p.R175G) not present in Pt 7's primary cells was detected in leukemic cells from one of the 3 individual xenografts (VAF: 51.8%). There was also one patient (Pt 4) with primary cells that did not harbor any founder mutations but contained 4 subclonal mutations (VAF: MYB, 6.9%; KRAS, 9.6%, PTPN11, 29.5%; ASXL1(p.T848fs*19), 35.6%). In the xenografts, the VAFs of MYB (0%) and PTPN11 (12.2%) were reduced, while the VAFs of the other 2 subclonal mutations were increased to 77.4% (KRAS) and 48.8% (ASXL1). These findings suggest that multiple genetically distinct LSC clone/subclones exist in patients with MPN-BP that are capable of engrafting NSG mice and have the potential in the future to participate in leukemia progression and/or relapse.
All 7 patients originally had a JAK2V617F+ MPN, but 3 of the MPN-BP cells were JAK2V617F¯. Among the remaining 4 patients whose primary cells retained JAK2V617F, the VAF was decreased in leukemic cells within the xenografts. These findings indicate that in JAK2V617F+ MPNs which evolve to MPN-BP, that the MPN-BP may either arise from stem cells distinct from the original JAK2V617F+ MPN stem cells with the acquisition of additional genetic events or progress from the clonal proliferation of distinct JAK2V617F¯ LSCs. Thus, JAK2V617F might play a limited role in either MPN-BP initiation or its progression. Four patients had distinct oncogenic TP53 mutations in primary cells which represented a clonal (VAF: Pt 2, 63.5%; Pt 5, 59.6%) or a subclonal mutation (Pts 1, 7) with a very low VAF (2.5%, 0.4%). The VAF for TP53 mutations were each increased in paired xenograft from 3 patients (Pt 2: 99.8%; Pt 1: 15.5%; Pt 7: 3.8%). These observations support a consistent proliferative advantage of LSCs carrying TP53 mutations in MPN-BP irrespective of how small the original LSC subclone is in the primary cells. Pt 3 and Pt 4 both had a KRAS mutation in their primary cells (VAF: Pt 3, 45.7%; Pt 4, 9.6%). For Pt 3, although the VAF of the KRAS mutation remained the same in the leukemic cells of the primary NSG recipient (44.4%), it increased to 90.2% in a secondary NSG recipient which was characterized by a higher leukemic cell burden (1.8-fold) and shorter survival (12 days less) than the primary NSG recipient. The VAF of KRAS mutation in Pt 4 also reached 77.4% in leukemic cells in the primary xenograft. These data suggest that acquisition of a KRAS mutation in LSCs may not only promote LSC clonal expansion, but also confer enhanced LSC self-renewal capacity. Finally, 4 distinct TET2 mutations were present as a founder mutation in primary cells of 3 patients but their VAF was unchanged or only slightly increased in leukemic cells in paired xenografts, suggesting that TET mutations are not involved in either LSC clonal expansion or disease progression. We conclude that TP53 and/or KRAS mutations appear to play an important role in the development of MPN-BP and may serve as potential targets for the development of effective treatment.
Rampal:Agios, Apexx, Blueprint Medicines, Celgene, Constellation, and Jazz: Consultancy; Constellation, Incyte, and Stemline Therapeutics: Research Funding. Hoffman:Merus: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal