

Background
Down Syndrome (DS) Acute Lymphoblastic Leukemia (ALL) patients have extremely poor outcomes with mortality rates four times greater than non-DS ALL patients within their first two years of diagnosis. They are more suspectible to treatment related toxicities and experience higher relapse rates compared to other ALL patients. Approximately 60% of DS-ALL patients harbor rearrangement of cytokine receptor like factor 2 (CRLF2r), specifically P2RY8-CRLF2, and/or the CRLF2 F232C activating mutation. These lesions are considered poor risk and currently no targeted therapy exist. How increased chromosome 21 gene dosage affect disease phenotype is not yet fully elucidated. However, the high mobility group nucleosome-binding domain-containing protein 1 (HMGN1) on chromosome 21, which competes with histone H1 to bind the nucleosome and results in gene activation may be a candidate for targeted therapy in DS-ALL.
Methods
We aimed to determine the role of HMGN1 in CRLF2r DS-ALL. To model CRLF2r DS-ALL, the trisomy 21 cell line, SET-2, was transduced with a retroviral vector encoding the CRLF2 F232C activating mutation. Gene knockdown of HMGN1 using CRISPR/Cas9 was performed in the SET-2 CRLF2r line and the non-trisomy-21, non-CRLF2 expressing Jurkat line. Individual knockdowns of another two genes on chromosome 21, DYRK1A and ERG were also performed. Knockdown of JAK2 was used as a control as it is critical for CRLF2 signaling. CellTiter-Glo was used to investigate proliferation of knockdown lines to test the hypothesis that HMGN1 is essential for CRLF2r DS-ALL cell proliferation.
Lentiviral vectors encoding the P2RY8-CRLF2 fusion gene, CRLF2 F232C activating mutation or an overexpression construct of HMGN1 were transduced into BaF3 cells individually or in combination to test the hypothesis that overexpressing HMGN1 is associated with activation of CRLF2. Quantitative PCR (qRTPCR) for CRLF2 and flow cytometry for the CRLF2/IL7Rα receptor (TSLPR) were used to determine the effect of increased HMGN1 on CRLF2 expression. AnnexinV/7-AAD cell death assays were performed to determine if the effects of HMGN1 could be reduced by the demethylase inhibitor GSK-J4.
Results
Knockdown of HMGN1 resulted in an 80-90% decrease in HMGN1 protein expression in SET-2 CRLF2 and Jurkat lines compared to the Cas9 controls. While knockdowns of DYRK1A and ERG did not impair the proliferation of SET-2 CRLF2 cells, HMGN1 and JAK2 knockdowns led to a complete proliferation arrest over a period of 120hrs (p=<0.001, n=3), demonstrating their effect on cell division. However, no change in proliferation was observed in the Jurkat knockdown lines.
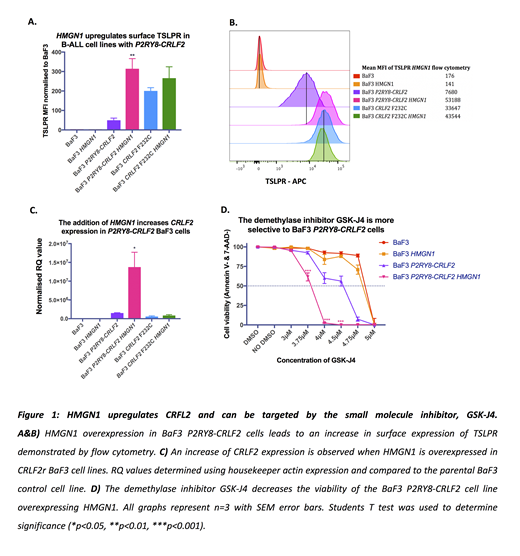
Overexpression of either HMGN1 or P2RY8-CRLF2 alone in BaF3 cells did not result in cytokine independent transformation. However, cytokine independence was triggered in BaF3 cells when HMGN1 and P2RY8-CRLF2 were co-expressed (p=<0.001, n=3); demonstrating a role for HMGN1 in leukemic transformation. Importantly, the overexpression of HMGN1 in the BaF3 P2RY8-CRLF2 line increased the mRNA expression of CRLF2 by 5.8-fold compared to the BaF3 P2RY8-CRLF2 line without HMGN1 (p=0.034, n=3) and increased the mean fluorescence intensity of TSLPR by flow cytometry from 42 to 308 (p=0.008, n=3) (figure 1.a-c) indicating a novel role for HMGN1 in P2RY8-CRLF2 activation.
While there are no pharmacological inhibitors for HMGN1, Lane et al. (2014) have shown that the restoration of H3K27 methylation using the demethylase inhibitor GSK-J4 was able to prevent DS-ALL cells from repassaging. Therefore, we have employed the inhibitor GSK-J4 to determine if it can reduce cell survival in HMGN1 overexpressed BaF3 cells. Specific inhibition of BaF3 P2RY8-CRLF2 HMGN1 cells was evident by decreased cell viability at a concentration of 3.8µM compared to BaF3 P2RY8-CRLF2 or BaF3 HMGN1 lines (p=<0.001, n=3) (figure 4.d). Thus, demonstrating a role for HMGN1 in the modification of the P2RY8-CRLF2 methylome and suggesting HMGN1 as a potential therapeutic target.
Conclusion
These data support the hypotheses that HMGN1 has a significant role in DS-ALL cell proliferation and that overexpression of HMGN1 results in activation of P2RY8-CRLF2. This is the first report of a novel role for HMGN1 in P2RY8-CRLF2 activation and leukemic transformation in CRLF2r DS-ALL. Additionally, we show that HMGN1 is a potential candidate for the development of a pharmacological inhibitor for CRLF2r DS-ALL.
Yeung:Novartis: Honoraria, Research Funding; BMS: Honoraria, Research Funding; Pfizer: Honoraria; Amgen: Honoraria. White:BMS: Honoraria, Research Funding; AMGEN: Honoraria, Speakers Bureau.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal