

Immune aplastic anemia (AA) is a life-threatening bone marrow failure syndrome in which the hematopoietic stem cells are destroyed, leading to pancytopenia. Although the exact biological process leading to AA remains largely unknown, bone marrow destruction is thought to be mediated by an autologous T cell response. We hypothesized that the autoimmune process in AA would create a T cell fingerprint unique to aplastic anemia.
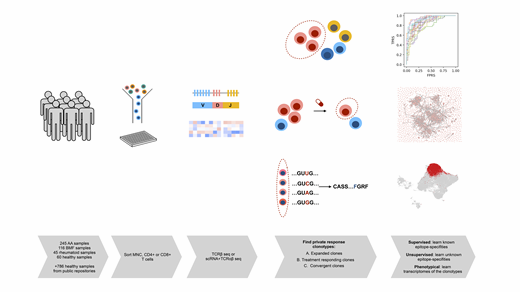
To decipher this signature, we collected an international, multi-centre cohort of 245 AA-samples from bone marrow and peripheral blood profiled with T-cell receptor beta (TCRβ) -sequencing. CD8+ T cell- and MNC-sorted samples from 153 clinically annotated AA patients were obtained from diagnosis, during remission and at relapse. To compare AA to similar diseases, we gathered 116 samples from other bone marrow failure syndromes, including MDS, PNH and hypoplastic LGL, and 45 samples from other autoimmune disorders. As healthy controls, we profiled 60 CD4+ and CD8+ T cells and utilized 786 MNC samples from public data repositories. To gain insight into T cell phenotypes, we also profiled 6 longitudinal samples with scRNA+TCRαβ-sequencing.
As there are 1x1012-16 different TCRs and most of them are exclusive to individuals (private), we reasoned that by studying the most biologically interesting clonotypes from each individual, we could explain differences in disease severities, variation in treatment responses and pathogenesis. From all subjects, we selected private response clonotypes: highly expanded clonotypes (at least 1% of the total repertoire), convergent clonotypes (in which multiple nucleotide sequences converge to encode the same amino acid sequence) and from patients with AA, treatment-responding clonotypes (clonotypes that were suppressed/expanded after immune therapy).
To analyse epitope-specificities of these clonotypes we leveraged TCRGP, our recently described Gaussian process method that can predict if TCRs recognize previously known epitopes. Clonotypes recognising viral epitopes (CMV, EBV and Influenza A) were enriched among private response clonotypes in comparison to the total repertoire (Fisher's exact test, p=2e-16), indicating that our filtering strategy indeed enriched for epitope-specific clonotypes. Of interest, the healthy donors' private response clonotypes showed more anti-viral clonotypes than did AA-patients (p=0.003), suggesting that in AA the epitope-specifities of private response clonotypes are not driven by common viral antigens.
To identify specifities against unknown epitopes of the private response AA clones, we used an unsupervised learning strategy, GLIPH,that groups TCRs recognising the same epitope based on amino acid level similarities. Clonotypes in AA showed high convergence in their epitope-targets, as 1709 of 5744 (29.75%) clonotypes formed a single, potentially epitope-specific cluster that was not viral-specific. Similar analysis of control samples resulted in fewer clones clustering to the most prominent cluster (23.20%, p=1.63e-10), suggesting for a more homogenous target population within AA patients' clones.
After showing sequence-level similarity of the private response clonotypes in AA, we aimed to link these pathological clonotypes to transcriptomes at the single-cell level using scRNA+TCRαβ-sequenced samples. The cells of the private response clonotypes showed multiple T cell phenotypes, but most cells (47.13%) in the bone marrow environment were recently activated CD8+ effector phenotype, marked by expression of GZMH, GNLY and PRF1. In comparison, the anti-viral clonotypes were mostly (37.3%) central memory phenotype (CCR7, TCF7). In serially sampled patients, anti-thymocyte globulin treatment suppressed private response clonotypes in a responding patient (55.03% of T cells to 12.79%), while the amount of these clonotypes increased in a non-responding patient (18.65% to 37.86%), where treatment mostly affected the viral-specific clonotypes.
In summary, our data suggest that the private response clonotypes in immune AA patients may recognise a common antigen, which was not predicted to be viral. Further, at the single-cell level AA signature clonotypes are of effector phenotype and fluctuate following immunosuppressive treatment. Monitoring of these clonotypes throughout treatment may provide insight into disease biology and variation in treatment responses.
Blombery:Janssen: Honoraria; Novartis: Consultancy; Invivoscribe: Honoraria. Maciejewski:Novartis: Consultancy; Alexion: Consultancy. Mustjoki:BMS: Honoraria, Research Funding; Novartis: Research Funding; Pfizer: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal