

Key Points
Cyfip1 plays a crucial role for branching of actin filaments and for lamellipodium formation.
Lamellipodium formation is not required for the formation of a hemostatic plug or thrombus.

Abstract
During platelet spreading, the actin cytoskeleton undergoes rapid rearrangement, forming filopodia and lamellipodia. Controversial data have been published on the role of lamellipodia in thrombus formation and stability. The Wiskott-Aldrich syndrome protein-family verprolin-homologous protein (WAVE)-regulatory complex, which has been shown in other cells to drive lamellipodium formation by enhancing actin nucleation via the actin-related protein 2/3 (Arp2/3) complex, is activated by Ras-related C3 botulinum toxin substrate 1 (Rac1) interaction with the WAVE complex subunit cytoplasmic fragile X mental retardation 1–interacting protein 1 (Cyfip1). We analyzed Cyfip1flox/floxPf4-Cre mice to investigate the role of Cyfip1 in platelet function. These mice displayed normal platelet counts and a slight reduction in platelet volume. Activation of mutant platelets was only moderately reduced to all tested agonists as measured by αIIbβ3 integrin activation and P-selectin surface exposure. However, lamellipodium formation of mutant platelets was completely abolished on different matrices. Nevertheless, Cyfip1-deficient platelets formed stable thrombi on collagen fibers ex vivo and in 2 models of occlusive arterial thrombosis in vivo. Similarly, the hemostatic function and maintenance of vascular integrity during inflammation of the skin and lung were unaltered in the mutant mice. Investigation of platelet morphology in an induced thrombus under flow revealed that platelets rather form filopodia in the thrombus shell, and are flattened with filopodium-like structures when in direct contact to collagen fibers at the bottom of the thrombus. We provide for the first time direct evidence that platelet lamellipodium formation is not required for stable thrombus formation, and that morphological changes of platelets differ between a static spreading assay and thrombus formation under flow.
Introduction
Circulating platelets are essential players in hemostasis and thrombosis. Upon vascular injury, platelets rapidly adhere to exposed extracellular matrix proteins and form a hemostatic plug that not only limits excessive blood loss, but also represents a major pathomechanism of ischemic cardio- and cerebrovascular diseases.1 Thrombus formation is a multistep process. At first, platelet tethering is mediated by the interaction of glycoprotein Ib (GPIb) with immobilized von Willebrand factor on exposed subendothelial collagen. This interaction enables binding of GPVI to collagen, which in turn initiates cellular activation. Subsequent inside-out upregulation of integrin affinity, synthesis of soluble mediators, granule release, and coagulant activity contributes to firm platelet adhesion and thrombus growth.2
The cytoskeleton ensures normal platelet size and shape, and plays a fundamental role for platelet function. Upon agonist-induced platelet activation, a rapid rearrangement of the cytoskeleton takes place, which results in platelet shape change and contributes to thrombus formation and stability.3 In vitro studies have shown that platelets getting in contact with immobilized adhesive proteins such as fibrinogen, start to spread and form first finger-like protrusions, so-called filopodia, which are finally remodeled to lamellipodia, which are flat, undulating cellular protrusions with actin-related protein 2/3 (Arp2/3)-mediated orthogonally arrayed short actin filaments. A fully spread platelet displays a circumferential zone of orthogonally arrayed short filaments within lamellipodia.4 Controversial data have been published on the role of lamellipodia in thrombus formation and stability, which were mainly based on inhibitor studies or on analyses of platelets with additional functional defects.5-9 A role for the small GTPase Ras-related C3 botulinum toxin substrate 1 (Rac1) in platelet lamellipodium formation was demonstrated using mice with deletion of Rac1 in the hematopoietic system (Mx1-Cre).5,6 However, although it was suggested that Rac1-regulated lamellipodium formation might be essential for maintaining integrity of platelet aggregates under shear,5 another study showed that Rac1 deficiency leads to a specific GPVI-dependent phospholipase Cγ2 (PLCγ2)-activation defect, resulting in impaired platelet adhesion and defective thrombus formation on collagen under flow. Supplementation with adenosine 5′-diphosphate (ADP) and a thromboxane A2 analog could rescue this defect.6 Furthermore, analysis of mice with a markedly decreased expression of the Arp2/3 complex9 revealed no significant defects in the hemostatic function or thrombus formation, although mutant platelets were unable to form lamellipodia. However, deletion of Arp2/3 complex subunit 2 (Arpc2) was not completely successful in megakaryocytes and platelets (>95% reduction),9 which could potentially rescue platelet function. Thus, the role of lamellipodia in this process is still unclear and direct proof has been lacking.
The Wiskott-Aldrich syndrome protein (WASp)-family verprolin-homologous protein (WAVE)-regulatory complex is a 5-subunit protein complex that includes the proteins WAVE-1/-2/-3, Abl interactor (ABI) 1/2, nucleosome assembly protein 1 (NAP1; also known as noncatalytic region of tyrosine kinase–associated protein 1 [NCKAP1]) or NCKAP1-like (NCKAP1L), cytoplasmic fragile X mental retardation 1–interacting protein 1/2 (Cyfip1/2), and hematopoietic stem/progenitor cell protein 300 (HSPC300).10 The WAVE complex is per se inactive, but is activated upon interaction of the subunit Cyfip1/2 and Rac1–guanosine triphosphate.11 The active WAVE complex interacts with Arp2/3, which in turn is crucial for the formation of branched actin filaments. WAVE-1 and -2 are the 2 major isoforms of WAVE in platelets.12 Homozygous disruption of the WAVE-1 gene resulted in postnatal lethality with an average lifespan of 23.6 days.13 Analysis of platelets of young mutant mice (age 14-20 days) revealed that WAVE-1 plays an important role downstream of GPVI.14 WAVE-2–null mice died at embryonic day 12.5 and their in vitro–differentiated megakaryocytes exhibited spreading defects.15 The Cyfip1 protein is mainly in a complex with the WAVE-2 isoform,16-18 however, its role for platelet function has not been studied.
We investigated the role of Cyfip1 in platelet physiology by generating Cyfip1fl/fl Pf4-Cre mice specifically lacking Cyfip1 in megakaryocytes and platelets. We demonstrate that Cyfip1fl/fl Pf4-Cre mice display normal platelet counts, and that activation of their platelets is only moderately decreased. However, mutant platelets are unable to form lamellipodia on different matrices, making this mouse line a unique model in which to study the function of platelet lamellipodia. We finally provide definite evidence that lamellipodium formation is dispensable for thrombus growth and stability, and that morphological changes of platelets differ between a static spreading assay and thrombus formation under flow.
Material and methods
Animals
Animal studies were approved by the district government of Lower Franconia (Bezirksregierung Unterfranken). Male and female mice were analyzed between 4 and 20 weeks of age. The Cyfip1 flox mouse strain was created from an embryonic stem (ES) cell clone (DEPD00521_7_E11) obtained from the Knockout Mouse Project (KOMP) Repository (www.komp.org). Following importation, the ES cells were cultured on a mouse embryonic fibroblast feeder layer. Correct targeting of Cyfip1 was confirmed on DNA extracted from the ES cell clone by polymerase chain reaction (PCR) on the 5′ side (CATTCTAACGTTCAAGATCGCAGACCAG and CACAACGGGT TCTTCTGTTAGTCC; 7.5 kb) and on the 3′ side (CATGTCTGGATCCGGGGGTACC GCGTCGAG and GATCTCCTGGTGCGGCATGCAAGC; 7.1 kb). The presence of the isolated 3′ loxP site was also confirmed by PCR (AAGCAGTCCGTGCTGAGATG and GAACCAAGCTCCCAGATTCC; 262 bp). Following confirmation of the genotyping of the targeted clone, mouse lines were derived by injection of ES cells into C57BL/6J blastocysts. After breeding of chimeras, germline offspring were identified by coat color, and the presence of the modified allele was confirmed with the 3′ loxP primers just described. Mice were subsequently crossed with a mouse line expressing Flpe (Tg(ACTFLPe)9205Dym) to delete the selectable marker by recombination at the flippase-recognition target (FRT) sites.19 Correct removal of the lacZ gene and Neo cassette was confirmed by PCR across the remaining FRT site (CTCTTCTTGTGGACCGCATC and CACGGTAGCCTGACAAGAGG; 562 bp). To study the function of Cyfip1 in platelets, Cyfip1fl/fl mice (supplemental Figure 1A, available on the Blood Web site) were intercrossed with mice carrying the Cre recombinase under the platelet factor 4 (Pf4) promoter20 to generate platelet- and megakaryocyte-specific Cyfip1-knockout mice. Cyfip1fl/fl Pf4-Cre is further referred to as Cyfip1−/−, Cyfip1fl/fl is further referred to as Cyfip1+/+, which served as controls.
Platelet preparation, immunoblotting, aggregometry, flow cytometry, platelet spreading, electron and superresolution microscopy, bleeding time, clot retraction, ex vivo and in vivo thrombus formation, reverse passive Arthus reaction, and lipopolysaccharide (LPS)-induced inflammation are described in supplemental Material and methods.
Data analysis
The presented results are mean plus or minus standard deviation from at least 2 to 3 independent experiments per group, if not stated otherwise. Differences between control and knockout mice were statistically analyzed using the Mann-Whitney U test. P values <.05 were considered as statistically significant: *P < .05; **P < .01; ***P < .001. Results with a P value >.05 were considered as not significant.
Results
Cyfip1−/− mice display normal platelet counts and only moderately impaired platelet activation
We confirmed lack of the Cyfip1 protein in platelet lysates of Cyfip1−/− mice by western blot analysis (Figure 1A; supplemental Figure 1B). WAVE-2 expression was reduced in mutant platelets, indicating instability of the complex in the absence of Cyfip1, however, expression of Rac1, Arpc2, and WAVE-1 remained unaltered (Figure 1A; supplemental Figure 1C). Cyfip1−/− mice displayed normal blood cell count parameters, including platelet count, and only a slight but significant reduction in platelet volume (Figure 1B-C; mean platelet volume: Cyfip1+/+, 5.11 ± 0.16 fL; Cyfip1−/−, 4.83 ± 0.07 fL; ***P < .001; supplemental Figure 1D). Expression of prominent glycoproteins and platelet ultrastructure were comparable between Cyfip1+/+ and Cyfip1−/− mice (supplemental Figures 1E and 2).
![Moderately reduced activation of Cyfip1−/−platelets. (A) Expression of indicated proteins in control and mutant platelets was assessed by western blot analysis. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading control (n = 3). (B) Platelet count per microliter and (C) platelet size given as mean forward scatter (FSC) was determined via flow cytometry (n = 4, representative for 4 independent experiments). Values are mean plus or minus standard deviation (SD); ***P < .001. (D) Flow cytometric determination of inside-out activation of the αIIbβ3 integrin (JON/A-phycoerythrin [PE] antibody) and (E) degranulation-dependent P-selectin exposure (fluorescein isothiocyanate [FITC]-labeled anti–P-selectin antibody) in response to the agonists ADP, U46619 (thromboxane analog), thrombin (Thr), CRP, convulxin (CVX), and rhodocytin (RC) (n = 5, representative for 4 independent experiments). Values are mean plus or minus SD; **P < .01, ***P < .001. (F) Washed platelets from Cyfip1+/+ and Cyfip1−/− mice were activated with the indicated agonists. Second-wave inhibitors: indomethacin 10 µM, EDTA, apyrase 2 U/mL (representative curve of at least 6 independent samples). (G) Determination of tyrosine phosphorylation after stimulation with 1 μg/mL CRP under stirring conditions (1000 rpm) at 37°C. Fifty-microliter aliquots were taken at the indicated time points. Samples were blotted on a polyvinylidene difluoride (PVDF) membrane and stained using the 4G10 antibody. GAPDH served as loading control (n = 3). MFI, mean fluorescence intensity.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/25/10.1182_blood.2019002105/7/m_bloodbld2019002105r1f1.png?Expires=1769148318&Signature=2SwPs6cdliTez7Ug-FJ~rodQqfXSFPtOcm3iGchcCBq912JjrirVuLM2X37PcfSvL~XU~cqby2LgvIZFVE4Bz1nh14gnlajlbuTUXmIvYgvZd~8rSWo~3cTNguDjUrGEwB63p6FkWUp5cu64WGLgcq-9rAOFvQwRBomgGZpVgoy97RIH28n~fmzmVeSvYTFkVfOadGDBkNykJeCFgJdPJ2wPBbidDbuOPDVW2P0eb2qQEUGDm48NJjg9hrxx5IkQlU3UiaGdLYr8OIYCxOJitu36FBfB4vj8yk~itPA8kLo1Tj4KJQ8GUcFMNDD~bPKqGfNvPIXi10W7kARJtnR-Fg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Moderately reduced activation of Cyfip1−/−platelets. (A) Expression of indicated proteins in control and mutant platelets was assessed by western blot analysis. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading control (n = 3). (B) Platelet count per microliter and (C) platelet size given as mean forward scatter (FSC) was determined via flow cytometry (n = 4, representative for 4 independent experiments). Values are mean plus or minus standard deviation (SD); ***P < .001. (D) Flow cytometric determination of inside-out activation of the αIIbβ3 integrin (JON/A-phycoerythrin [PE] antibody) and (E) degranulation-dependent P-selectin exposure (fluorescein isothiocyanate [FITC]-labeled anti–P-selectin antibody) in response to the agonists ADP, U46619 (thromboxane analog), thrombin (Thr), CRP, convulxin (CVX), and rhodocytin (RC) (n = 5, representative for 4 independent experiments). Values are mean plus or minus SD; **P < .01, ***P < .001. (F) Washed platelets from Cyfip1+/+ and Cyfip1−/− mice were activated with the indicated agonists. Second-wave inhibitors: indomethacin 10 µM, EDTA, apyrase 2 U/mL (representative curve of at least 6 independent samples). (G) Determination of tyrosine phosphorylation after stimulation with 1 μg/mL CRP under stirring conditions (1000 rpm) at 37°C. Fifty-microliter aliquots were taken at the indicated time points. Samples were blotted on a polyvinylidene difluoride (PVDF) membrane and stained using the 4G10 antibody. GAPDH served as loading control (n = 3). MFI, mean fluorescence intensity.
Moderately reduced activation of Cyfip1−/−platelets. (A) Expression of indicated proteins in control and mutant platelets was assessed by western blot analysis. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading control (n = 3). (B) Platelet count per microliter and (C) platelet size given as mean forward scatter (FSC) was determined via flow cytometry (n = 4, representative for 4 independent experiments). Values are mean plus or minus standard deviation (SD); ***P < .001. (D) Flow cytometric determination of inside-out activation of the αIIbβ3 integrin (JON/A-phycoerythrin [PE] antibody) and (E) degranulation-dependent P-selectin exposure (fluorescein isothiocyanate [FITC]-labeled anti–P-selectin antibody) in response to the agonists ADP, U46619 (thromboxane analog), thrombin (Thr), CRP, convulxin (CVX), and rhodocytin (RC) (n = 5, representative for 4 independent experiments). Values are mean plus or minus SD; **P < .01, ***P < .001. (F) Washed platelets from Cyfip1+/+ and Cyfip1−/− mice were activated with the indicated agonists. Second-wave inhibitors: indomethacin 10 µM, EDTA, apyrase 2 U/mL (representative curve of at least 6 independent samples). (G) Determination of tyrosine phosphorylation after stimulation with 1 μg/mL CRP under stirring conditions (1000 rpm) at 37°C. Fifty-microliter aliquots were taken at the indicated time points. Samples were blotted on a polyvinylidene difluoride (PVDF) membrane and stained using the 4G10 antibody. GAPDH served as loading control (n = 3). MFI, mean fluorescence intensity.
Activation of mutant platelets was only moderately reduced in response to all tested agonists as measured by αIIbβ3 integrin activation and P-selectin surface exposure (Figure 1D-E). To test the functional consequences of this moderate activation defect, we performed in vitro aggregation studies. Cyfip1−/− platelets aggregated normally to the G protein–coupled agonist thrombin (Figure 1F) and the GPVI-specific agonist collagen-related peptide (CRP) even at very low concentrations (Figure 1F). We only consistently observed a delay in onset of aggregation in response to collagen. However, the amplitude as a measure for maximum platelet aggregation was comparable between control and mutant platelets (Figure 1F). Next, we preincubated washed platelets with apyrase (2 U/mL) and indomethacin (10 μM) to prevent the effect of second-wave mediators, and then activated with high concentrations of collagen. We found that Cyfip1−/− platelets aggregated normally at high collagen concentrations under these conditions (10 and 20 µg/mL collagen; Figure 1F), excluding a selective GPVI-signaling defect in Cyfip1−/− platelets as observed for Rac1-deficient platelets.6 Next, we assessed changes in protein tyrosine phosphorylation upon stimulation with CRP and detected a comparable total tyrosine phosphorylation pattern in Cyfip1−/− platelets (Figure 1G).
The Arp2/3 complex is activated by the WAVE-regulatory complex downstream of Rac1. Therefore, we performed activation studies with control platelets in the presence of the Arp2/3 inhibitor, CK-666, and the fungal toxin, cytochalasin D, a potent actin polymerization inhibitor. Control platelets incubated with CK-666 (50 µM) displayed reduced activation similar to the reduction seen with Cyfip1−/− platelets (αIIbβ3 activation: supplemental Figure 3A; P-selectin exposure: supplemental Figure 3B). Preincubation of platelets with cytochalasin D had a stronger effect on agonist-induced activation as compared with Cyfip1-deficient platelets (supplemental Figure 3). These data suggest that the moderately reduced activation of Cyfip1−/− platelets is rather caused by an Arp2/3-related defect than an upstream signaling defect. Taken together, lack of Cyfip1 only moderately and nonselectively impairs platelet activation, which has no major effect on platelet-platelet interaction.
Absent lamellipodium formation of Cyfip1−/− platelets on different matrices in static spreading assays
It has been shown that the WAVE-regulatory complex drives lamellipodium formation in other cell types.21-23 This prompted us to study the role of the WAVE complex subunit Cyfip1 in platelet lamellipodium formation. Four consecutive stages of platelet spreading can be observed: (I) adhesion of resting platelets, (II) formation of filopodia, (III) a combination of filopodia and lamellipodia, and (IV) only lamellipodia. Cyfip1−/− platelets adhered to fibrinogen under static conditions, formed filopodia, but strikingly, lamellipodia formation (phases 3 and 4) was completely abolished (Figure 2A-B; supplemental Videos 1 and 2). However, the transition from resting platelet to filopodia-forming platelet was unaltered between both groups as revealed by live imaging of platelet spreading (Figure 2C). At the 30-minute time point, spread mutant platelets were smaller but had an increased perimeter, and formed more filopodia with increased length (Figure 2D-G). To compare the defect of lamellipodium formation of Cyfip1−/− platelets to reagents interfering with actin branching or polymerization, we incubated control platelets with different concentrations of CK-666 and cytochalasin D prior spreading, and observed that a concentration of 50 µM CK-666 or higher resulted in a significant reduction of fully spread platelets (phase 4), however, even at 200 µM CK-666 ∼30% of platelets could be observed in phase 3 (supplemental Figure 4A). Nevertheless, these data show that the spreading defect of CK-666–treated platelets resembles to a large extent the phenotype of the Cyfip1-deficient platelets. In contrast, platelets were unable to change their shape when actin polymerization was inhibited by cytochalasin D (supplemental Figure 4B). These results suggest that Cyfip1-deficient platelets cannot form Arp2/3-dependent branched actin filaments, which are necessary for lamellipodium formation.
![Cyfip1-deficient platelets are unable to form lamellipodia on a fibrinogen-coated surface. (A) Washed platelets were allowed to spread on fibrinogen, following fixation at the indicated time points (n = 3, representative for 3 independent experiments). Scale bar, 5 µm. (B) Quantification of the different spreading phases of fixed platelets after 5, 15, and 30 minutes from differential interference contrast images (Zeiss Axiovert 200 inverted microscope [100×/1.4 oil objective]). (C) Phase abundance of live cells over time (wild type [WT], n = 58; knockout [KO], n = 32). Quantification of (D) platelet size and (E) platelet perimeter (WT, n = 100; KO, n = 58) of all Cyfip1+/+ and Cyfip1−/− platelets present in a visual field after 30-minute spreading. (F) Length and (G) number of filopodia (WT, n = 58; KO, n = 32) of live platelets in phase 2 spreading. Values are mean plus or minus SD. ***P < .001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/25/10.1182_blood.2019002105/7/m_bloodbld2019002105r1f2.png?Expires=1769148318&Signature=YzAbjF9~ymEqSqhbBhfQDxRKszyEc9cSSDndAi5WrWSuvp6edPV6lr9UkC08pKuGnD9bDygxdQuuoc83pZfZrFJRHIP~920EdkPzD71FD9K2xgnWHxcZNKbM0gUxSqPS6MSrNS~Vo7m24UnFjxjbC3ZOAsXzvwr9r2Bt8Q4yOV6JMXYNrlWAjZZWNB2m2N4WfNZbRhcOx8-CDUZV8as46dJKRxk0PM~IqB7N0BSP3MlE3ovu4MHjWjWokJ-ZKCYCunmOF~4n1qQ4MIw7WaPDEW~RTq~4~q60DI2kEAnjd78PG~d3IUPs-lhmxaZqMuSczLa7BMYcK24kXJzhHv19Vg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Cyfip1-deficient platelets are unable to form lamellipodia on a fibrinogen-coated surface. (A) Washed platelets were allowed to spread on fibrinogen, following fixation at the indicated time points (n = 3, representative for 3 independent experiments). Scale bar, 5 µm. (B) Quantification of the different spreading phases of fixed platelets after 5, 15, and 30 minutes from differential interference contrast images (Zeiss Axiovert 200 inverted microscope [100×/1.4 oil objective]). (C) Phase abundance of live cells over time (wild type [WT], n = 58; knockout [KO], n = 32). Quantification of (D) platelet size and (E) platelet perimeter (WT, n = 100; KO, n = 58) of all Cyfip1+/+ and Cyfip1−/− platelets present in a visual field after 30-minute spreading. (F) Length and (G) number of filopodia (WT, n = 58; KO, n = 32) of live platelets in phase 2 spreading. Values are mean plus or minus SD. ***P < .001.
Cyfip1-deficient platelets are unable to form lamellipodia on a fibrinogen-coated surface. (A) Washed platelets were allowed to spread on fibrinogen, following fixation at the indicated time points (n = 3, representative for 3 independent experiments). Scale bar, 5 µm. (B) Quantification of the different spreading phases of fixed platelets after 5, 15, and 30 minutes from differential interference contrast images (Zeiss Axiovert 200 inverted microscope [100×/1.4 oil objective]). (C) Phase abundance of live cells over time (wild type [WT], n = 58; knockout [KO], n = 32). Quantification of (D) platelet size and (E) platelet perimeter (WT, n = 100; KO, n = 58) of all Cyfip1+/+ and Cyfip1−/− platelets present in a visual field after 30-minute spreading. (F) Length and (G) number of filopodia (WT, n = 58; KO, n = 32) of live platelets in phase 2 spreading. Values are mean plus or minus SD. ***P < .001.
Therefore, we investigated the localization of Arp2/3 in spread platelets and found that Arp2/3 was strongly expressed at the circumferential zone of control platelets (supplemental Figure 5). Interestingly, Arp2/3 also localized to filopodia in control and mutant platelets, indicating that not altered expression or localization, but impaired Cyfip1-mediated Arp2/3 activation is causative for abolished lamellipodium formation. To assess this in more detail, we analyzed the cytoskeletal organization of spread platelets by platinum replica electron24 (Figure 3A) and superresolution microscopy by direct stochastic optical reconstruction microscopy (dSTORM)25 (supplemental Figure 6). Orthogonally arrayed short actin filaments were formed in the circumferential zone of spread control platelets. In contrast, Cyfip1−/− platelets showed only filopodia consisting of parallel bundles of actin filaments (Figure 3A). In agreement, Cyfip1−/− platelets were principally able to assemble filamentous actin as we determined the F-actin content in resting platelets and in platelets after activation with CRP or thrombin by flow cytometry. Only slightly decreased content was observed in CRP-activated platelets but not resting or thrombin-activated platelets (Figure 3B). However, the F-actin ratio of activated platelets over resting platelets revealed no significant differences, demonstrating that mutant platelets can assemble F-actin after activation similar to controls (Figure 3B). Next, we tested whether Cyfip1-deficient platelets form lamellipodia on other surfaces. Likewise, mutant platelets could not form lamellipodia on collagen IV, laminin, and CRP and were unable to reorganize actin filaments into short, branched filaments (Figure 4).
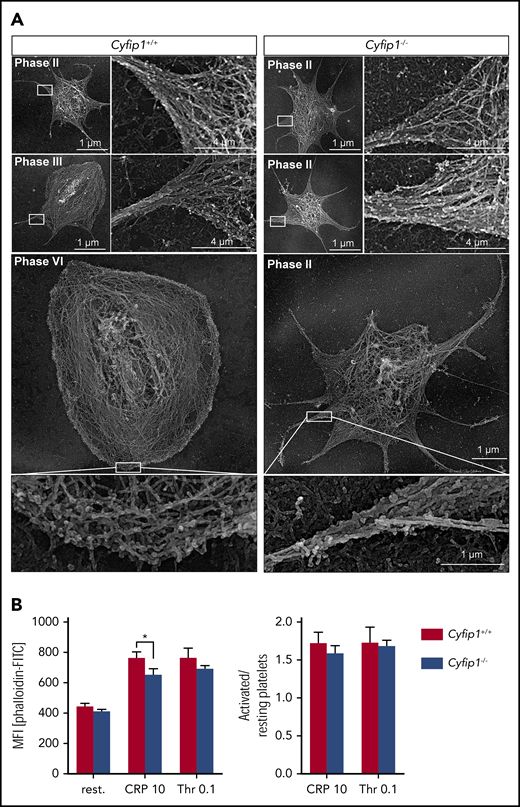
Cyfip1-deficient platelets cannot form branched actin filaments. (A) Platinum replica electron microscopic images of the cytoskeleton ultrastructure of platelets spread on fibrinogen after 15 minutes. Scale bars, 1 or 4 µm. (B) Left, F-actin content under resting conditions and upon stimulation with CRP (10 μg/ml) or thrombin (0.1 U/ml) was assessed by measuring the MFI of bound FITC-labeled phalloidin in flow cytometry. Right, Ratio of MFI of activated vs resting samples reflecting F-actin polymerization rates (n = 4, representative for 3 independent experiments). Values are mean plus or minus SD. *P < .05.
Cyfip1-deficient platelets cannot form branched actin filaments. (A) Platinum replica electron microscopic images of the cytoskeleton ultrastructure of platelets spread on fibrinogen after 15 minutes. Scale bars, 1 or 4 µm. (B) Left, F-actin content under resting conditions and upon stimulation with CRP (10 μg/ml) or thrombin (0.1 U/ml) was assessed by measuring the MFI of bound FITC-labeled phalloidin in flow cytometry. Right, Ratio of MFI of activated vs resting samples reflecting F-actin polymerization rates (n = 4, representative for 3 independent experiments). Values are mean plus or minus SD. *P < .05.
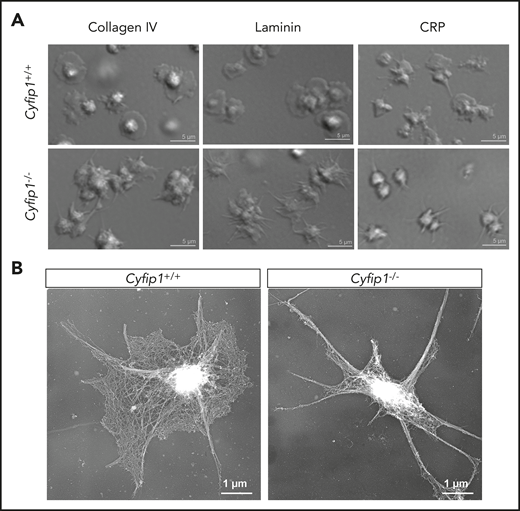
Abolished lamellipodium formation of mutant platelets on different matrices. (A) Washed platelets were allowed to spread on collagen IV, laminin, or CRP for 30 minutes (n = 3). Scale bars, 5 µm. (B) Images of the platelet cytoskeleton ultrastructure on CRP after 15 minutes (n = 5). Scale bars, 1 µm.
Abolished lamellipodium formation of mutant platelets on different matrices. (A) Washed platelets were allowed to spread on collagen IV, laminin, or CRP for 30 minutes (n = 3). Scale bars, 5 µm. (B) Images of the platelet cytoskeleton ultrastructure on CRP after 15 minutes (n = 5). Scale bars, 1 µm.
Lamellipodia are not required for ex vivo thrombus formation
Next, we tested the role of lamellipodia in thrombus formation in an in vitro system. Therefore, we perfused whole blood over a collagen-coated surface in a flow chamber at different shear rates (150 s−1, 1000 s−1, 1700 s−1). Interestingly, surface coverage (Figure 5A-B) and thrombus volume (Figure 5A,C) were comparable between samples from control and mutant mice (supplemental Videos 3 and 4). Kinetics of single-platelet adhesion and embolization of smaller platelet aggregates appeared normal. Similarly, Cyfip1−/− platelets showed effective thrombus compaction under flow (supplemental Video 4) and clot retraction in vitro (Figure 5D-E). Video analyses revealed that single platelets at times formed filopodia to grab collagen fibers, before they became activated and initiated thrombus formation (supplemental Video 5). Surprisingly, we have never observed fully spread platelets (equivalent to phase 4 platelets in static spreading assay) with circumferential lamellipodia under these conditions. These findings were corroborated when we removed the flow chamber from the glass coverslip after the experiment and prepared the sample for scanning (Figure 6A) and platinum replica (Figure 6B) electron microscopic analyses. Investigation of control and Cyfip1−/− platelet morphology showed that in general, platelets form filopodia in the thrombus shell (Figure 6A) and are flattened with filopodial structures and parallel actin bundles when in direct contact to collagen fibers at the bottom of the thrombus (Figure 6; supplemental Figure 7A-B), contrary to the abundant fully spread platelets with circumferential lamellipodia observed in a static spreading assay (Figures 2 and 3). Some unspread platelets at collagen fibers were found in control and mutant samples and only in a few control platelets could we identify small parts of branched actin filaments at the cell boundary (supplemental Figure 7A-B). Taken together, these results demonstrate that lamellipodium formation is not required for stable thrombus formation ex vivo and that morphological changes of platelets differ between a static spreading assay and thrombus formation under flow.
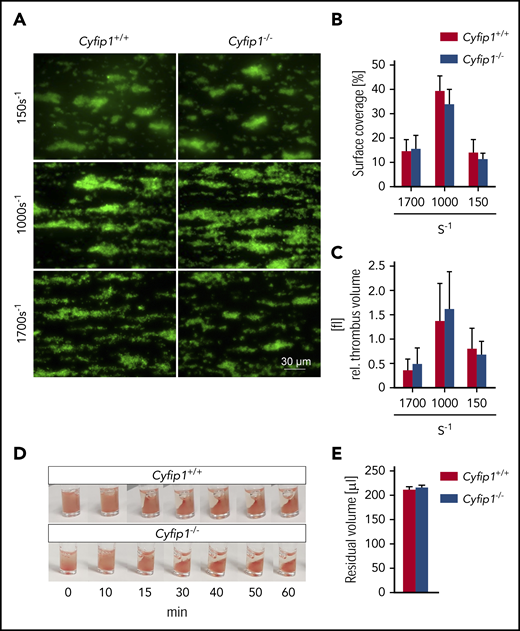
Normal ex vivo thrombus formation and clot retraction of Cyfip1-deficient platelets. (A) Thrombus formation was assessed on collagen at different shear rates. Shown are representative fluorescence pictures of platelets stained with a Dylight-488 anti-GPIX antibody (n = 5, representative for 3 independent experiments). Scale bar, 30 µm. (B) Surface coverage and (C) relative thrombus volume were quantified. (D) Clot formation was observed over time and (E) residual serum volume was measured (n = 3, representative for 2 independent experiments).
Normal ex vivo thrombus formation and clot retraction of Cyfip1-deficient platelets. (A) Thrombus formation was assessed on collagen at different shear rates. Shown are representative fluorescence pictures of platelets stained with a Dylight-488 anti-GPIX antibody (n = 5, representative for 3 independent experiments). Scale bar, 30 µm. (B) Surface coverage and (C) relative thrombus volume were quantified. (D) Clot formation was observed over time and (E) residual serum volume was measured (n = 3, representative for 2 independent experiments).
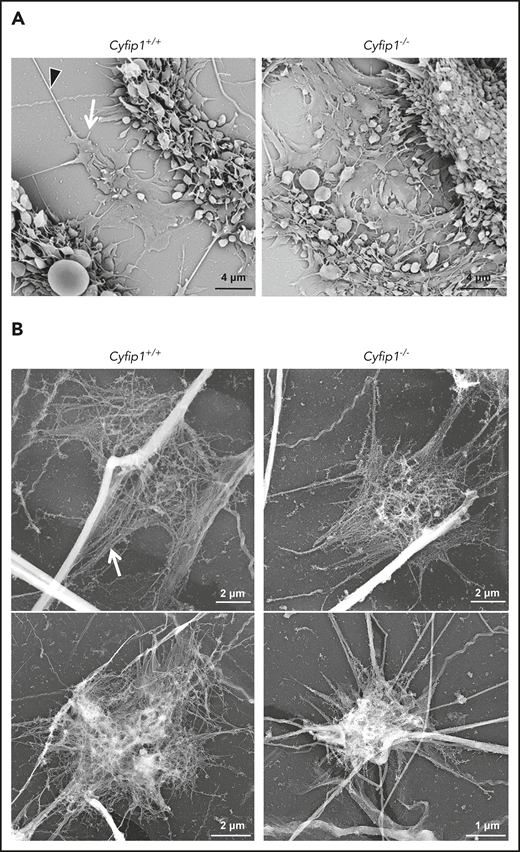
Flattened platelets with filopodium-like structures at the bottom of the thrombus. (A) Scanning electron microscopic image of platelets after experimentally induced ex vivo thrombus formation on collagen. White arrow indicates a filopodium; black arrowhead indicates collagen fiber. Scale bar, 4 µm. (B) Platinum replica of platelets after experimentally induced ex vivo thrombus formation on collagen. White arrow indicates a filopodium with unbranched actin filaments. Scale bars, 1 and 2 µm.
Flattened platelets with filopodium-like structures at the bottom of the thrombus. (A) Scanning electron microscopic image of platelets after experimentally induced ex vivo thrombus formation on collagen. White arrow indicates a filopodium; black arrowhead indicates collagen fiber. Scale bar, 4 µm. (B) Platinum replica of platelets after experimentally induced ex vivo thrombus formation on collagen. White arrow indicates a filopodium with unbranched actin filaments. Scale bars, 1 and 2 µm.
Cyfip1−/− platelets can expose phosphatidylserine on the surface
A distinct population of highly activated platelets has been described residing close to collagen fibers and as patches around a thrombus. These platelets are characterized by surface‐exposed phosphatidylserine (PS), which supports coagulation factor complex assembly, and hence is a key regulatory event in murine arterial thrombus formation.26,27 To test whether Cyfip1−/− platelets can regulate coagulant activity, washed platelets were stimulated with ionomycin, a combination of CRP and thrombin or alone, and PS exposure was determined by annexin V binding. Although ionomycin efficiently induced PS exposure, platelets stimulated with thrombin or CRP alone could hardly bind annexin V. The combination of CRP and thrombin induced PS exposure on >40% of control and mutant platelets (supplemental Figure 8A-B). Next, we perfused whole blood over immobilized collagen and observed that the number and surface coverage of PS+ collagen-adherent platelets were comparable between control and mutant samples (supplemental Figure 8C-D). These data show that platelets can regulate coagulant activity without the necessity to form lamellipodia.
Normal bleeding times, arterial thrombus formation, and maintenance of vascular integrity in Cyfip1−/− mice
Next, we analyzed the relevance of lamellipodial structures for in vivo platelet function. Bleeding times after tail-tip amputation were comparable between control and Cyfip1−/− mice (Figure 7A). The application of 20% FeCl3 on the exteriorized mesenteric arterioles of Cyfip1−/− mice resulted in comparable platelet adhesion, appearance of first thrombi (Figure 7B-C), and time to formation of occlusive thrombi (Figure 7D; supplemental Videos 6 and 7). Similarly, thrombus growth was studied in a second model of arterial thrombosis, in which the abdominal aorta is mechanically injured and blood flow is monitored by an ultrasonic perivascular Doppler flowmeter. In this model, thrombus formation and occlusion of the vessel in Cyfip1−/− mice occurred with similar kinetics as in controls (Figure 7E; supplemental Figure 9A). Finally, we isolated the aorta and analyzed the shape of single platelets in the experimentally induced in vivo thrombus (Figure 7F; supplemental Figure 9B). Again, we could not observe platelets with lamellipodia, but rather platelets that formed filopodia. This shows that lamellipodial structures are not required for classical formation of a hemostatic plug or a pathological thrombus in vivo.
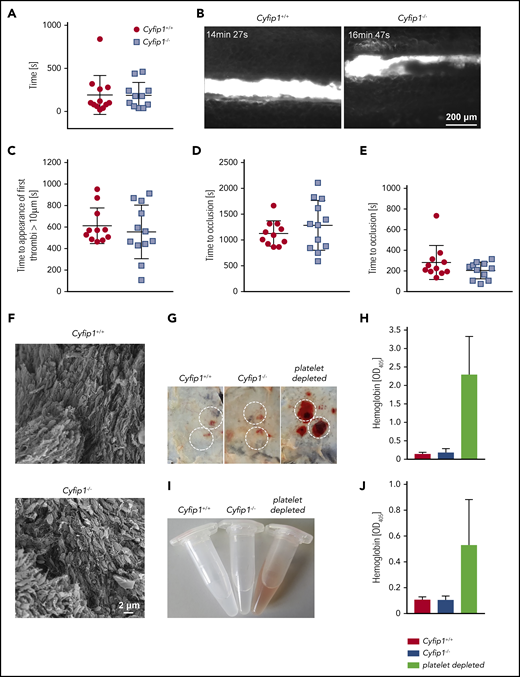
Lamellipodium formation is not required for hemostatic function and arterial thrombus formation. (A) A 2-mm segment of the tail tip was cut, and bleeding was determined to have ceased when no blood drop was observed on the filter paper. Each symbol represents 1 individual. (B) Mesenteric arterioles were treated with 20% FeCl3, and adhesion and thrombus formation of fluorescently labeled platelets were monitored by in vivo fluorescence microscopy. Representative images are shown. (C) Statistical evaluation of the time to appearance of a first thrombus and (D) time to occlusion. (E) The abdominal aorta was mechanically injured using forceps (compression for 15 seconds), and blood flow was monitored. Each symbol represents 1 individual. (F) Scanning electron microscopy of an in vivo formed thrombus (n = 4). Scale bar, 2 µm. (G-H) Mice were subjected to the rpA reaction to induce local skin inflammation. (G) Representative images are shown. Inflammatory spots are highlighted. Original image: 4 × 3 cm. (H) Hemoglobin (Hb) content in tissue punch biopsies from inflammatory spots was quantified (n = 4, representative for 3 independent experiments). (I) Representative images of bronchoalveolar lavage liquid at 4 hours after LPS application. (J) Quantification of the Hb content in bronchoalveolar lavage liquid. Platelet-depleted animals served as positive control of inflammatory bleeding in both models (n = 5, representative for 3 independent experiments).
Lamellipodium formation is not required for hemostatic function and arterial thrombus formation. (A) A 2-mm segment of the tail tip was cut, and bleeding was determined to have ceased when no blood drop was observed on the filter paper. Each symbol represents 1 individual. (B) Mesenteric arterioles were treated with 20% FeCl3, and adhesion and thrombus formation of fluorescently labeled platelets were monitored by in vivo fluorescence microscopy. Representative images are shown. (C) Statistical evaluation of the time to appearance of a first thrombus and (D) time to occlusion. (E) The abdominal aorta was mechanically injured using forceps (compression for 15 seconds), and blood flow was monitored. Each symbol represents 1 individual. (F) Scanning electron microscopy of an in vivo formed thrombus (n = 4). Scale bar, 2 µm. (G-H) Mice were subjected to the rpA reaction to induce local skin inflammation. (G) Representative images are shown. Inflammatory spots are highlighted. Original image: 4 × 3 cm. (H) Hemoglobin (Hb) content in tissue punch biopsies from inflammatory spots was quantified (n = 4, representative for 3 independent experiments). (I) Representative images of bronchoalveolar lavage liquid at 4 hours after LPS application. (J) Quantification of the Hb content in bronchoalveolar lavage liquid. Platelet-depleted animals served as positive control of inflammatory bleeding in both models (n = 5, representative for 3 independent experiments).
It has been reported that inflammation represents an unconventional hemostatic situation in which individual platelets are sufficient to prevent bleeding.28 To test whether platelet lamellipodial structures play a role in this process, we challenged mice in 2 different models, namely the reverse passive Arthus reaction (IV injection of antigen, followed by intradermal injection of antibody) and LPS-induced inflammation. As expected, when we depleted platelets in mice by injection of an anti-GPIbα antibody, we observed severe inflammatory bleeding in the skin (Figure 7G-H) and into the lung (Figure 7I-J), demonstrating the importance of platelets to maintain vascular integrity as reported previously.29 Finally, control and Cyfip1−/− mice were subjected to the reverse passive Arthus reaction of the skin and LPS-induced lung inflammation. Cyfip1−/− mice did not show signs of bleeding in these experimental settings, suggesting that platelet lamellipodium formation is dispensable for maintaining vascular integrity at sites of acute inflammation (Figure 7G-J).
Discussion
Static platelet spreading assays, particularly on fibrinogen-coated surfaces, are frequently performed as a measure of the contribution of outside-in signaling and cytoskeletal rearrangement in platelet function30 as well as readout for functional properties of in vitro–produced platelets.31-34 In vitro, 4 stages of platelet spreading can be observed: (I) adhesion of resting platelets, (II) formation of finger-like protrusions, called filopodia, (III) a combination of filopodia and plate-like extensions, called lamellipodia, and (IV) only lamellipodia. Under dynamic conditions, platelets adhere to the exposed extracellular matrix, become activated, and thereby change their shape, which is accompanied by the secretion and synthesis of several prothrombotic factors, which together contribute to the activation of more platelets and finally lead to aggregate formation.
Whether the stages of in vitro platelet spreading, particularly lamellipodium formation, also contribute to platelet adhesion and activation under dynamic conditions in vivo has been debated for years. Platelet lamellipodia have been suggested to be important for the initiation of thrombus growth when platelets are in direct contact with extracellular matrix components, most notably collagen.5,7,8 In contrast, other publications provided indirect evidence that lamellipodium formation is not crucial for thrombus formation and hemostasis.6,9,35 However, the mouse models of Rac1 and Arp2/3 deficiency displayed severe defects in GPVI signaling6 and platelet production as well as knockout efficiency,9 respectively. In this study, we provide for the first time definitive evidence that platelet lamellipodium formation is not required for stable thrombus formation ex vivo and in vivo, hemostatic plug formation, as well as for maintenance of vascular integrity in inflammatory settings (Figures 5-7) by analyzing Cyfip1-deficient mice with abolished lamellipodium formation but only minimal side effects in platelet function (Figures 1-4). Moreover, we demonstrate by direct observation that morphological changes of platelets differ between a static spreading assay and thrombus formation under flow, where platelets only formed filopodia to adhere to collagen fibers (supplemental Video 5) and to form a stable thrombus as visualized by scanning electron microscopy in an experimentally induced arterial thrombus (Figure 7F). We could further show that control and mutant platelets in direct contact with collagen fibers at the bottom of the thrombus were flattened but formed filopodial structures with parallel actin filaments. Fully spread platelets with circumferential lamellipodia, as can be observed in the static spreading assay (phase 4), were absent under these conditions (Figure 6; supplemental Figure 7). However, although platelet lamellipodia are not required for the formation and overall stability of thrombi, we cannot exclude the possibility that lamellipodium formation might still be important in other thrombi-related processes, such as vessel repair.
Interestingly, it was previously demonstrated that Arp2/3 complex activation is normal in platelets lacking WASp and hypothesized that other proteins, probably WAVE-2 or cortactin or both, activate the Arp2/3 complex.36 We could show that Cyfip1-deficient platelets have a reduced WAVE-2 expression (Figure 1A) and cannot form Arp2/3-dependent branched actin filaments (Figure 3A). Thus, our data strongly suggest for the first time that the WAVE-2 protein/complex regulates Arp2/3 activity in platelets.
It was recently shown that collagen-adherent platelets can transform into PS-exposing balloon-like structures, which was termed procoagulant spreading and likely contributes to hemostatic and thrombotic responses in vivo by amplifying procoagulant responses.37 We tested the ability of mutant platelets to expose PS on the surface after stimulation with CRP and thrombin in vitro and under dynamic conditions ex vivo, and obtained comparable results for control and mutant mice (supplemental Figure 8). Therefore, we suggest that platelets do not need to form lamellipodia in order to expose PS on the surface. Hence, the bridging mechanism of platelet and coagulation response seems to be intact in Cyfip1-deficient platelets, enabling them to initiate thrombus formation.
However, 1 open question remains: what is the role of lamellipodia in platelet function? There are several possibilities: (I) lamellipodial structures could be an in vitro phenomenon during static platelet spreading. However, this is rather unlikely, because many other cells also produce this dense network of branched actin filaments contributing to important cellular functions. (II) The platelet machinery to form lamellipodia might be a relic of evolution no longer needed for the specialized functions platelets have, and are thus dispensable. Until now, we cannot exclude either possibility, however, it is most likely that (III) platelet lamellipodia play an important role in other processes besides the initiation of thrombus formation and thrombus stability. These membrane protrusions at the leading edge of cells drive cell migration in many physiological and pathological situations.38 Recently, it was reported that platelets actively migrate at sites of a formed thrombus.39 The authors concluded from their findings that platelet migration might be too slow to contribute to thrombus formation, but might play a role in thrombus reorganization and consolidation.39 Our data of the Cyfip1−/− mice show that platelet lamellipodia, which are most likely a prerequisite for platelet migration, are not important for thrombus formation, supporting the conclusion by Gaertner et al of the dispensable role of platelet migration in thrombus growth. We did not directly analyze thrombus reorganization, however, we visualized single platelets in an experimentally induced arterial thrombus and found that platelets exclusively form filopodia or filopodium-like structures (Figure 7F). Therefore, we suggest that at least lamellipodia are not important in the reorganization of a thrombus. Interestingly, Gaertner et al could show that platelets also migrate to sites of infection in order to help trap bacteria and clear the vascular surface.39 The authors used platelets from Myh9−/− mice, which are unable to migrate,39 however, can extend lamellipodia but without stress fiber–like formation.40 It will be interesting to investigate whether Cyfip1-deficient platelets with absent lamellipodium formation can migrate in vitro and in vivo, and whether Cyfip1 is a key protein in bacteria scavenging and bundling at sites of infection.
In summary, we show that Cyfip1 deficiency does not alter platelet biogenesis and has no major impact on platelet activation, but Cyfip1 plays a crucial role for branching of actin filaments and consequently for lamellipodium formation in vitro. We took advantage of this unique mouse model and could demonstrate that lamellipodium formation is not required for the formation of a hemostatic plug or a thrombus. Moreover, Cyfip1-deficient mice could maintain vascular integrity at the site of inflammation. Finally, we suggest that megakaryocyte-/platelet-specific Cyfip1-deficient mice represent a unique mouse system in which to study the role of platelet lamellipodia in other platelet-dependent processes in vivo. Further studies will be required to address this.
The data generated and analyzed in this study are available from the corresponding author on reasonable request.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Daniela Naumann for excellent technical assistance.
This work was supported by an Emmy Noether grant (BE5084/3-1 [M.B.]) and TR240 grant with project number 374031971 (A06 [M.B.], and A07 [B.N. and M.S.]) of the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation), and by Cancer Research UK core grant numbers A17196 and A15673 [L.M.].
Authorship
Contribution: Y.S. performed experiments, analyzed data, and wrote the manuscript; A.S., S. Beck, J.V., L.R., C.K., and P.E. performed experiments and analyzed data; S. Bryson generated the mice and analyzed data; M.S. and B.N. analyzed data; L.M. analyzed data and contributed to the writing of the manuscript; and M.B. supervised research, performed experiments, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Markus Bender, Institute of Experimental Biomedicine, Chair I, University Hospital Würzburg, Josef-Schneider-Str 2, 97080 Würzburg, Germany; e-mail: bender_m1@ukw.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal