

Key Points
NPM1 mutations in AML arise from replication errors primed by illegitimate TdT activity.
The involvement of TdT in both NPM1 and FLT3-ITD mutagenesis suggests a significant proportion of AML is a by-product of adaptive immunity.

Abstract
Nucleophosmin (NPM1) is the most commonly mutated gene in acute myeloid leukemia (AML). AML with mutated NPM1 is recognized as a separate entity in the World Health Organization 2016 classification and carries a relatively favorable prognosis. NPM1 mutations are predominantly 4-bp duplications or insertions in the terminal exon that arise through an unknown mechanism. Here we analyze 2430 NPM1 mutations from 2329 adult and 101 pediatric patients to address their origin. We show that NPM1 mutations display the hallmarks of replication slippage, but lack suitable germline microhomology available for priming. Insertion mutations display G/C-rich N-nucleotide tracts, with a significant bias toward polypurine and polypyrimidine stacking (P < .001). These features suggest terminal deoxynucleotidyl transferase (TdT) primes replication slippage through N-nucleotide addition, with longer syntheses manifesting as N-regions. The recurrent type A, type D, and type B mutations require 1, 2, and 3 N-nucleotide extensions of T, CC, and CAT, respectively, with the last nucleotide used as occult microhomology. This TdT-mutator model successfully predicts the relative incidence of the 256 potential 4-bp insertion/duplication mutations at position c.863_864 over 4 orders of magnitude (ρ = 0.484, P < .0001). Children have a different NPM1 mutation spectrum to adults, including a shift away from type A mutations and toward longer N-regions, consistent with higher TdT activity in pediatric myeloid stem cells. These findings complement our FLT3-ITD data, suggesting illegitimate TdT activity contributes to around one-half of AMLs. AML may therefore reflect the price for adaptive immunity.
Introduction
Nucleophosmin (NPM1) mutations drive 33% of cases of acute myeloid leukemia (AML), including >50% of cases with a normal karyotype.1,2 NPM1-mutated AML is regarded as a distinct entity in the World Health Organization 2016 classification of myeloid neoplasms3 on the basis of exclusivity with recurrent cytogenetic rearrangements, CD34 negativity, multilineage involvement, and gene expression profile.4-8 NPM1 mutations predict a favorable prognosis unless a FLT3-ITD is also present9,10 ; hence, NPM1 and FLT3-ITD mutation status is routinely determined at presentation to facilitate risk-adapted therapy.
NPM1 is a nucleolar protein that shuttles between nucleus and cytoplasm, contributing to multiple cellular processes.5,6 In AML, mutated NPM1 is relocated from the nucleoli to the cytoplasm2 by creation of a C-terminal leucine-rich nuclear export signal (NES),11 most commonly of the form L-xxx-V-xx-V-x-L.5,6 Disruption of the existing nucleolar localization signal through mutation of tryptophans 288 and 290, or just W290, is also required to create a transforming protein.6,12,13 Loss of W290 alone necessitates creation of a stronger NES, with substitution of the second hydrophobic position valine with leucine, phenylalanine, cysteine, or methionine (eg, L-xxx-L-xx-V-x-L).13 Relocation of the mutated protein to the cytoplasm is essential for transformation.14
At the DNA level, these changes are achieved through frame-shift of the terminal coding exon. The most common NPM1 mutation (type A, incidence ∼80%)6 is a 4-bp duplication, c.860_863dupTCTG,2 which creates an L-xxx-V-xx-V-x-L NES with loss of both W288 and W290.5,6 Type B (c.863_864insCATG) and type D (c.863_864insCCTG) mutations represent ∼10% and ∼5% of cases, respectively. All other mutations are considerably rarer,6 including other 4-bp insertions at c.863_864, and longer insertions (± deletions) at nearby positions, such as c.869_873delins(9) which retains W288 and creates a stronger NES.
It is unknown how NPM1 mutations arise, although a replicative origin is plausible because the most common mutation is a duplication.15 However, there is a significant difference in the incidence of type A mutations between children and adults, potentially suggesting different pathological mechanisms.5,6,16,17 Any model should account for this difference. In our accompanying paper, we propose a replication-based model to explain the genesis of FLT3-ITDs in which terminal deoxynucleotidyl transferase (TdT) synthesizes the microhomology for priming replication slippage when germline microhomology is unavailable, with longer syntheses manifesting as G/C-rich N-regions at the repeat junction.18 The marked cooccurrence of NPM1 and FLT3-ITD mutations suggested to us that TdT could also prime NPM1 mutations (although these mutations are also known to synergize19 ).
Here we analyze 2430 NPM1 mutations to explore their genesis. NPM1 mutations show anatomy consistent with TdT-primed replication slippage, including G/C-rich N-regions with polypurine/polypyrimidine base stacking. Modeling with TdT predicts the frequency of all 256 different 4-bp insertions at c.863_864, and explains the different NPM1 mutational spectra observed in children and adults. These observations strengthen the evidence that TdT causes leukemia.
Materials and methods
Mutation identification
NPM1 mutation sequences were identified from papers published up to 2016 via PubMed (supplemental Table 1, available on the Blood Web site). Duplicate publications were excluded. The NPM1 reference sequence was LRG_458t1, numbering from the start of the coding region.
NPM1 mutation dinucleotide analysis
Dinucleotides were identified from NPM1 N-regions ≥2 nt. All insertion positions were considered. Each individual mutation was only included once irrespective of incidence.
Incidence calculations
The predicted incidence of each NPM1 4-bp c.863_864 insertion was calculated using the incidences of the 16 TdT-synthesized dinucleotides and the proportion of each TdT extension length, as determined from FLT3-ITD N-regions (supplemental Table 2), and corrected for the percentage of 1-5 nucleotide extensions from c.863 predicted to yield a transforming protein (6.7%). Probabilities were summed for each extension length through which a mutation could occur, excluding nucleotide extensions ≥6 nt.
Bioinformatics and statistics
Human codon usage was obtained from http://www.kazusa.or.jp/codon/. Cumulative binomial probabilities were calculated at http://vassarstats.net/binomialX.html. Fisher’s exact tests were performed at http://www.graphpad.com/quickcalcs/contingency1/. P values are 2-tailed. Spearman’s rank correlations were performed at http://www.wessa.net/rwasp_spearman.wasp/.
Results
NPM1 mutation anatomy and priming
The position, sequence, and incidence of 2423 NPM1 mutations (2322 adults, 101 children) was compiled (Tables 1 and2). Seven additional unique adult NPM1 mutations of unknown incidence were appended (Table 1). We identified 114 unique mutations, predominantly short frameshift duplications, insertions, or indels that impaired the nucleolar localization signal and created an NES.
Adult NPM1 mutation sequences
| Type | Nucleotide sequence of NPM1 mutations | Translation | Occurrence |
|---|---|---|---|
| WT | gatctctggcagtggaggaagtctctttaa | DLWQWRKSL | NA |
| gatct ATGC ctggcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 1 | |
| gatct TTGC ctggcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 1 | |
| gatct TTGTCTG gcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 1 | |
| gatctc ACAA tggcagtggaggaagtctctttaagaaaatag | DLTMAVEEVSLRK | 1 | |
| gatctc ATTAA ggcagtggaggaagtctctttaagaaaatag | DLIKAVEEVSLRK | 1 | |
| gatctc ATTC tggcagtggaggaagtctctttaagaaaatag | DLILAVEEVSLRK | 1 | |
| gatctc CGCA tggcagtggaggaagtctctttaagaaaatag | DLRMAVEEVSLRK | 1 | |
| gatctc GTTTTGGCGA tggaggaagtctctttaagaaaatag | DLVLAMEEVSLRK | 1 | |
| gatctc TCCATGCTCC tggaggaagtctctttaagaaaatag | DLSMLLEEVSLRK | 1 | |
| gatctct CCCG ggcagtggaggaagtctctttaagaaaatag | DLSRAVEEVSLRK | 1 | |
| gatctct CTGCAGCCT tggaggaagtctctttaagaaaatag | DLSAALEEVSLRK | 1 | |
| gatctctg CAAA gcagtggaggaagtctctttaagaaaatag | DLCKAVEEVSLRK | 1 | |
| gatctctg CAAG gcagtggaggaagtctctttaagaaaatag | DLCKAVEEVSLRK | 3 | |
| gatctctg CACA gcagtggaggaagtctctttaagaaaatag | DLCTAVEEVSLRK | 1 | |
| gatctctg CACCACCT tggaggaagtctctttaagaaaatag | DLCTTLEEVSLRK | 1 | |
| gatctctg CACG gcagtggaggaagtctctttaagaaaatag | DLCTAVEEVSLRK | 1 | |
| gatctctg CACGCG agtggaggaagtctctttaagaaaatag | DLCTRVEEVSLRK | 1 | |
| gatctctg CAGA gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 6 | |
| gatctctg CAGG gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 7 | |
| gatctctg CAGTAAG gtggaggaagtctctttaagaaaatag | DLCSKVEEVSLRK | 1 | |
| gatctctg CAGTCG agtggaggaagtctctttaagaaaatag | DLCSRVEEVSLRK | 1 | |
| B | gatctctg CATG gcagtggaggaagtctctttaagaaaatag | DLCMAVEEVSLRK | 197 |
| gatctctg CCAC gcagtggaggaagtctctttaagaaaatag | DLCHAVEEVSLRK | 1 | |
| gatctctg CCAG gcagtggaggaagtctctttaagaaaatag | DLCQAVEEVSLRK | 11 | |
| gatctctg CCAGAG agtggaggaagtctctttaagaaaatag | DLCQRVEEVSLRK | 1 | |
| gatctctg CCGA gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 3 | |
| gatctctg CCGC gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 1 | |
| gatctctg CCGCCT agtggaggaagtctctttaagaaaatag | DLCRLVEEVSLRK | 1 | |
| gatctctg CCGG gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 11 | |
| gatctctg CCGTT cagtggaggaagtctctttaagaaaatag | DLCRSVEEVSLRK | 1 | |
| D | gatctctg CCTG gcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 160 |
| gatctctg CGCC gcagtggaggaagtctctttaagaaaatag | DLCAAVEEVSLRK | 1 | |
| gatctctg CGTG gcagtggaggaagtctctttaagaaaatag | DLCVAVEEVSLRK | 3 | |
| gatctctg CTCG gcagtggaggaagtctctttaagaaaatag | DLCSAVEEVSLRK | 4 | |
| gatctctg CTGG gcagtggaggaagtctctttaagaaaatag | DLCWAVEEVSLRK | 1 | |
| gatctctg CTTG gcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 32 | |
| gatctctg TAAA gcagtggaggaagtctctttaagaaaatag | DLCKAVEEVSLRK | 1 | |
| gatctctg TAAG gcagtggaggaagtctctttaagaaaatag | DLCKAVEEVSLRK | 7 | |
| gatctctg TACCTTCC tggaggaagtctctttaagaaaatag | DLCTFLEEVSLRK | 1 | |
| gatctctg TACG gcagtggaggaagtctctttaagaaaatag | DLCTAVEEVSLRK | 1 | |
| gatctctg TAGC gcagtggaggaagtctctttaagaaaatag | DLCSAVEEVSLRK | 1 | |
| gatctctg TAGG gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 4 | |
| gatctctg TATG gcagtggaggaagtctctttaagaaaatag | DLCMAVEEVSLRK | 18 | |
| gatctctg TCAAGACTTTCTTA aagtctctttaagaaaatag | DLCQDFLKVSLRK | 1 | |
| gatctctg TCAG gcagtggaggaagtctctttaagaaaatag | DLCQAVEEVSLRK | 9 | |
| gatctctg TCAT gcagtggaggaagtctctttaagaaaatag | DLCHAVEEVSLRK | 2 | |
| gatctctg TCGC gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 1 | |
| gatctctg TCGG gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 8 | |
| gatctctg TCGGAGTCTCGGCGGAC tctctttaagaaaatag | DLCRSLGGLSLRK | 1 | |
| gatctctg TCGT gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 1 | |
| A | gatctctg TCTG gcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 1750 |
| gatctctg TGAC gcagtggaggaagtctctttaagaaaatag | DLCDAVEEVSLRK | 1 | |
| gatctctg TGCG gcagtggaggaagtctctttaagaaaatag | DLCAAVEEVSLRK | 1 | |
| gatctctg TGTG gcagtggaggaagtctctttaagaaaatag | DLCVAVEEVSLRK | 2 | |
| gatctctg TTCC gcagtggaggaagtctctttaagaaaatag | DLCSAVEEVSLRK | 1 | |
| gatctctg TTCG gcagtggaggaagtctctttaagaaaatag | DLCSAVEEVSLRK | 3 | |
| gatctctg TTTG gcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 2 | |
| gatctctggca AAGGA tggaggaagtctctttaagaaaatag | DLWQRMEEVSLRK | 1 | |
| gatctctggca AGATTTCTTAAATC gtctctttaagaaaa tagtttaa | DLWQDFLNRLFKK IV | 1 | |
| gatctctggcag AAGT tggaggaagtctctttaagaaaatag | DLWQKLEEVSLRK | 1 | |
| gatctctggcag AGAA tggaggaagtctctttaagaaaatag | DLWQRMEEVSLRK | 3 | |
| gatctctggcag AGAC tggaggaagtctctttaagaaaatag | DLWQRLEEVSLRK | 2 | |
| gatctctggcag AGAT tggaggaagtctctttaagaaaatag | DLWQRLEEVSLRK | 1 | |
| gatctctggcag AGGA tggaggaagtctctttaagaaaatag | DLWQRMEEVSLRK | 8 | |
| gatctctggcag AGGC tggaggaagtctctttaagaaaatag | DLWQRLEEVSLRK | 2 | |
| gatctctggcag CGCCTT gaggaagtctctttaagaaaatag | DLWQRLEEVSLRK | 1 | |
| gatctctggcag CGCT tggaggaagtctctttaagaaaatag | DLWQRLEEVSLRK | 1 | |
| gatctctggcag CGGA tggaggaagtctctttaagaaaatag | DLWQRMEEVSLRK | 1 | |
| gatctctggcag CGGATGGC ggaagtctctttaagaaaatag | DLWQRMAEVSLRK | 1 | |
| gatctctggcag CGGC tggaggaagtctctttaagaaaatag | DLWQRLEEVSLRK | 1 | |
| gatctctggcag CGTCTTGGCC aagtctctttaagaaaatag | DLWQRLGQVSLRK | 1 | |
| gatctctggcag CGTTTCC aggaagtctctttaagaaaatag | DLWQRFQEVSLRK | 1 | |
| gatctctggcag GGGATAGCGATGC tctctttaagaaaatag | DLWQGIAMLSLRK | 1 | |
| gatctctggcag GGGGTGGGGAATC tctctttaagaaaatag | DLWQGVGNLSLRK | 1 | |
| gatctctggcagt CCAT ggaggaagtctctttaagaaaatag | DLWQSMEEVSLRK | 1 | |
| gatctctggcagt CCCTAGCCC aagtctctttaagaaaatag | DLWQSLAQVSLRK | 1 | |
| gatctctggcagt CCCTCTCCC aagtctctttaagaaaatag | DLWQSLSQVSLRK | 1 | |
| gatctctggcagt CCCTGGAGA aagtctctttaagaaaatag | DLWQSLEKVSLRK | 1 | |
| gatctctggcagt CCCTTTCCA aagtctctttaagaaaatag | DLWQSLSKVSLRK | 1 | |
| gatctctggcagt CCCTTTCTA aagtctctttaagaaaatag | DLWQSLSKVSLRK | 1 | |
| gatctctggcagt CTCTTGCCC aagtctctttaagaaaatag | DLWQSLAQVSLRK | 1 | |
| gatctctggcagt CTCTTTCTA aagtctctttaagaaaatag | DLWQSLSKVSLRK | 1 | |
| gatctctggcagt CTTTCGCTCAC gtctctttaagaaaatag | DLWQSFAHVSLRK | 1 | |
| gatctctggcagt TATTTTCCC aagtctctttaagaaaatag | DLWQLFSQVSLRK | 1 | |
| gatctctggcagtg CCTCGAGA aagtctctttaagaaaatag | DLWQCLEKVSLRK | 1 | |
| gatctctggcagtg CTGCTCCC aagtctctttaagaaaatag | DLWQCCSQVSLRK | 1 | |
| gatctctggcagtg TTTCTCCC aagtctctttaagaaaatag | DLWQCFSQVSLRK | 1 | |
| gatctctggcagtg TTTTGCTC aagtctctttaagaaaatag | DLWQCFAQVSLRK | 1 | |
| gatctctggcagtg TTTTTCCC aagtctctttaagaaaatag | DLWQCFSQVSLRK | 2 | |
| gatctctg CAGGCT agtggaggaagtctctttaagaaaatag | DLCRLVEEVSLRK | Rare | |
| gatctctg CCGCGG agtggaggaagtctctttaagaaaatag | DLCRGVEEVSLRK | Rare | |
| gatctctg TAGGAAG gtggaggaagtctctttaagaaaatag | DLCRKVEEVSLRK | Rare | |
| gatctctggca AAGAA tggaggaagtctctttaagaaaatag | DLWQRMEEVSLRK | Rare | |
| gatctctggca CCGTTTCTCC gaagtctctttaagaaaatag | DLWHRFSEVSLRK | Rare | |
| gatctctggcag ACTTTCTATA aagtctctttaagaaaatag | DLWQTFYKVSLRK | Rare | |
| gatctctggcagtg CTTCTCCA aagtctctttaagaaaatag | DLWQCFSKVSLRK | Rare |
| Type | Nucleotide sequence of NPM1 mutations | Translation | Occurrence |
|---|---|---|---|
| WT | gatctctggcagtggaggaagtctctttaa | DLWQWRKSL | NA |
| gatct ATGC ctggcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 1 | |
| gatct TTGC ctggcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 1 | |
| gatct TTGTCTG gcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 1 | |
| gatctc ACAA tggcagtggaggaagtctctttaagaaaatag | DLTMAVEEVSLRK | 1 | |
| gatctc ATTAA ggcagtggaggaagtctctttaagaaaatag | DLIKAVEEVSLRK | 1 | |
| gatctc ATTC tggcagtggaggaagtctctttaagaaaatag | DLILAVEEVSLRK | 1 | |
| gatctc CGCA tggcagtggaggaagtctctttaagaaaatag | DLRMAVEEVSLRK | 1 | |
| gatctc GTTTTGGCGA tggaggaagtctctttaagaaaatag | DLVLAMEEVSLRK | 1 | |
| gatctc TCCATGCTCC tggaggaagtctctttaagaaaatag | DLSMLLEEVSLRK | 1 | |
| gatctct CCCG ggcagtggaggaagtctctttaagaaaatag | DLSRAVEEVSLRK | 1 | |
| gatctct CTGCAGCCT tggaggaagtctctttaagaaaatag | DLSAALEEVSLRK | 1 | |
| gatctctg CAAA gcagtggaggaagtctctttaagaaaatag | DLCKAVEEVSLRK | 1 | |
| gatctctg CAAG gcagtggaggaagtctctttaagaaaatag | DLCKAVEEVSLRK | 3 | |
| gatctctg CACA gcagtggaggaagtctctttaagaaaatag | DLCTAVEEVSLRK | 1 | |
| gatctctg CACCACCT tggaggaagtctctttaagaaaatag | DLCTTLEEVSLRK | 1 | |
| gatctctg CACG gcagtggaggaagtctctttaagaaaatag | DLCTAVEEVSLRK | 1 | |
| gatctctg CACGCG agtggaggaagtctctttaagaaaatag | DLCTRVEEVSLRK | 1 | |
| gatctctg CAGA gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 6 | |
| gatctctg CAGG gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 7 | |
| gatctctg CAGTAAG gtggaggaagtctctttaagaaaatag | DLCSKVEEVSLRK | 1 | |
| gatctctg CAGTCG agtggaggaagtctctttaagaaaatag | DLCSRVEEVSLRK | 1 | |
| B | gatctctg CATG gcagtggaggaagtctctttaagaaaatag | DLCMAVEEVSLRK | 197 |
| gatctctg CCAC gcagtggaggaagtctctttaagaaaatag | DLCHAVEEVSLRK | 1 | |
| gatctctg CCAG gcagtggaggaagtctctttaagaaaatag | DLCQAVEEVSLRK | 11 | |
| gatctctg CCAGAG agtggaggaagtctctttaagaaaatag | DLCQRVEEVSLRK | 1 | |
| gatctctg CCGA gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 3 | |
| gatctctg CCGC gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 1 | |
| gatctctg CCGCCT agtggaggaagtctctttaagaaaatag | DLCRLVEEVSLRK | 1 | |
| gatctctg CCGG gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 11 | |
| gatctctg CCGTT cagtggaggaagtctctttaagaaaatag | DLCRSVEEVSLRK | 1 | |
| D | gatctctg CCTG gcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 160 |
| gatctctg CGCC gcagtggaggaagtctctttaagaaaatag | DLCAAVEEVSLRK | 1 | |
| gatctctg CGTG gcagtggaggaagtctctttaagaaaatag | DLCVAVEEVSLRK | 3 | |
| gatctctg CTCG gcagtggaggaagtctctttaagaaaatag | DLCSAVEEVSLRK | 4 | |
| gatctctg CTGG gcagtggaggaagtctctttaagaaaatag | DLCWAVEEVSLRK | 1 | |
| gatctctg CTTG gcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 32 | |
| gatctctg TAAA gcagtggaggaagtctctttaagaaaatag | DLCKAVEEVSLRK | 1 | |
| gatctctg TAAG gcagtggaggaagtctctttaagaaaatag | DLCKAVEEVSLRK | 7 | |
| gatctctg TACCTTCC tggaggaagtctctttaagaaaatag | DLCTFLEEVSLRK | 1 | |
| gatctctg TACG gcagtggaggaagtctctttaagaaaatag | DLCTAVEEVSLRK | 1 | |
| gatctctg TAGC gcagtggaggaagtctctttaagaaaatag | DLCSAVEEVSLRK | 1 | |
| gatctctg TAGG gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 4 | |
| gatctctg TATG gcagtggaggaagtctctttaagaaaatag | DLCMAVEEVSLRK | 18 | |
| gatctctg TCAAGACTTTCTTA aagtctctttaagaaaatag | DLCQDFLKVSLRK | 1 | |
| gatctctg TCAG gcagtggaggaagtctctttaagaaaatag | DLCQAVEEVSLRK | 9 | |
| gatctctg TCAT gcagtggaggaagtctctttaagaaaatag | DLCHAVEEVSLRK | 2 | |
| gatctctg TCGC gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 1 | |
| gatctctg TCGG gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 8 | |
| gatctctg TCGGAGTCTCGGCGGAC tctctttaagaaaatag | DLCRSLGGLSLRK | 1 | |
| gatctctg TCGT gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 1 | |
| A | gatctctg TCTG gcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 1750 |
| gatctctg TGAC gcagtggaggaagtctctttaagaaaatag | DLCDAVEEVSLRK | 1 | |
| gatctctg TGCG gcagtggaggaagtctctttaagaaaatag | DLCAAVEEVSLRK | 1 | |
| gatctctg TGTG gcagtggaggaagtctctttaagaaaatag | DLCVAVEEVSLRK | 2 | |
| gatctctg TTCC gcagtggaggaagtctctttaagaaaatag | DLCSAVEEVSLRK | 1 | |
| gatctctg TTCG gcagtggaggaagtctctttaagaaaatag | DLCSAVEEVSLRK | 3 | |
| gatctctg TTTG gcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 2 | |
| gatctctggca AAGGA tggaggaagtctctttaagaaaatag | DLWQRMEEVSLRK | 1 | |
| gatctctggca AGATTTCTTAAATC gtctctttaagaaaa tagtttaa | DLWQDFLNRLFKK IV | 1 | |
| gatctctggcag AAGT tggaggaagtctctttaagaaaatag | DLWQKLEEVSLRK | 1 | |
| gatctctggcag AGAA tggaggaagtctctttaagaaaatag | DLWQRMEEVSLRK | 3 | |
| gatctctggcag AGAC tggaggaagtctctttaagaaaatag | DLWQRLEEVSLRK | 2 | |
| gatctctggcag AGAT tggaggaagtctctttaagaaaatag | DLWQRLEEVSLRK | 1 | |
| gatctctggcag AGGA tggaggaagtctctttaagaaaatag | DLWQRMEEVSLRK | 8 | |
| gatctctggcag AGGC tggaggaagtctctttaagaaaatag | DLWQRLEEVSLRK | 2 | |
| gatctctggcag CGCCTT gaggaagtctctttaagaaaatag | DLWQRLEEVSLRK | 1 | |
| gatctctggcag CGCT tggaggaagtctctttaagaaaatag | DLWQRLEEVSLRK | 1 | |
| gatctctggcag CGGA tggaggaagtctctttaagaaaatag | DLWQRMEEVSLRK | 1 | |
| gatctctggcag CGGATGGC ggaagtctctttaagaaaatag | DLWQRMAEVSLRK | 1 | |
| gatctctggcag CGGC tggaggaagtctctttaagaaaatag | DLWQRLEEVSLRK | 1 | |
| gatctctggcag CGTCTTGGCC aagtctctttaagaaaatag | DLWQRLGQVSLRK | 1 | |
| gatctctggcag CGTTTCC aggaagtctctttaagaaaatag | DLWQRFQEVSLRK | 1 | |
| gatctctggcag GGGATAGCGATGC tctctttaagaaaatag | DLWQGIAMLSLRK | 1 | |
| gatctctggcag GGGGTGGGGAATC tctctttaagaaaatag | DLWQGVGNLSLRK | 1 | |
| gatctctggcagt CCAT ggaggaagtctctttaagaaaatag | DLWQSMEEVSLRK | 1 | |
| gatctctggcagt CCCTAGCCC aagtctctttaagaaaatag | DLWQSLAQVSLRK | 1 | |
| gatctctggcagt CCCTCTCCC aagtctctttaagaaaatag | DLWQSLSQVSLRK | 1 | |
| gatctctggcagt CCCTGGAGA aagtctctttaagaaaatag | DLWQSLEKVSLRK | 1 | |
| gatctctggcagt CCCTTTCCA aagtctctttaagaaaatag | DLWQSLSKVSLRK | 1 | |
| gatctctggcagt CCCTTTCTA aagtctctttaagaaaatag | DLWQSLSKVSLRK | 1 | |
| gatctctggcagt CTCTTGCCC aagtctctttaagaaaatag | DLWQSLAQVSLRK | 1 | |
| gatctctggcagt CTCTTTCTA aagtctctttaagaaaatag | DLWQSLSKVSLRK | 1 | |
| gatctctggcagt CTTTCGCTCAC gtctctttaagaaaatag | DLWQSFAHVSLRK | 1 | |
| gatctctggcagt TATTTTCCC aagtctctttaagaaaatag | DLWQLFSQVSLRK | 1 | |
| gatctctggcagtg CCTCGAGA aagtctctttaagaaaatag | DLWQCLEKVSLRK | 1 | |
| gatctctggcagtg CTGCTCCC aagtctctttaagaaaatag | DLWQCCSQVSLRK | 1 | |
| gatctctggcagtg TTTCTCCC aagtctctttaagaaaatag | DLWQCFSQVSLRK | 1 | |
| gatctctggcagtg TTTTGCTC aagtctctttaagaaaatag | DLWQCFAQVSLRK | 1 | |
| gatctctggcagtg TTTTTCCC aagtctctttaagaaaatag | DLWQCFSQVSLRK | 2 | |
| gatctctg CAGGCT agtggaggaagtctctttaagaaaatag | DLCRLVEEVSLRK | Rare | |
| gatctctg CCGCGG agtggaggaagtctctttaagaaaatag | DLCRGVEEVSLRK | Rare | |
| gatctctg TAGGAAG gtggaggaagtctctttaagaaaatag | DLCRKVEEVSLRK | Rare | |
| gatctctggca AAGAA tggaggaagtctctttaagaaaatag | DLWQRMEEVSLRK | Rare | |
| gatctctggca CCGTTTCTCC gaagtctctttaagaaaatag | DLWHRFSEVSLRK | Rare | |
| gatctctggcag ACTTTCTATA aagtctctttaagaaaatag | DLWQTFYKVSLRK | Rare | |
| gatctctggcagtg CTTCTCCA aagtctctttaagaaaatag | DLWQCFSKVSLRK | Rare |
Inserted bases are shown in capital letters in the second column. The 4 conserved hydrophobic positions of the nuclear export signal are in bold-face type in the third column. The first base shown is c.856 from the coding reference sequence.
NA, not available.
Pediatric NPM1 Mutation Sequences
| Type | Nucleotide sequence of NPM1 mutations | Translation | Occurrence |
|---|---|---|---|
| WT | gatctctggcagtggaggaagtctctttaa | DLWQWRKSL | NA |
| gatctct ATCT ggcagtggaggaagtctctttaagaaaatag | DLYLAVEEVSLRK | 1 | |
| gatctctg CAGG gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 1 | |
| B | gatctctg CATG gcagtggaggaagtctctttaagaaaatag | DLCMAVEEVSLRK | 18 |
| gatctctg CCGG gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 3 | |
| D | gatctctg CCTG gcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 8 |
| gatctctg CGGA gcagtggaggaagtctctttaagaaaatag | DLCGAVEEVSLRK | 1 | |
| gatctctg CTTG gcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 1 | |
| gatctctg TACG gcagtggaggaagtctctttaagaaaatag | DLCTAVEEVSLRK | 1 | |
| gatctctg TATG gcagtggaggaagtctctttaagaaaatag | DLCMAVEEVSLRK | 1 | |
| gatctctg TCAG gcagtggaggaagtctctttaagaaaatag | DLCQAVEEVSLRK | 1 | |
| gatctctg TCGG gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 1 | |
| A | gatctctg TCTG gcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 43 |
| gatctctg TGCC gcagtggaggaagtctctttaagaaaatag | DLCAAVEEVSLRK | 1 | |
| gatctctg TGTG gcagtggaggaagtctctttaagaaaatag | DLCVAVEEVSLRK | 2 | |
| gatctctggc TCCGATTTGC ggaagtctctttaagaaaatag | DLWLRFAEVSLRK | 1 | |
| gatctctggca AGATCTCAGCAAG gtctctttaagaaaatag | DLWQDLSKVSLRK | 1 | |
| gatctctggcag CGGA tggaggaagtctctttaagaaaatag | DLWQRMEEVSLRK | 1 | |
| gatctctggcag CGGATGGCC gaagtctctttaagaaaatag | DLWQRMAEVSLRK | 1 | |
| gatctctggcag CGGATTCC ggaagtctctttaagaaaatag | DLWQRIPEVSLRK | 2 | |
| gatctctggcag CGTTC ggaggaagtctctttaagaaaatag | DLWQRSEEVSLRK | 1 | |
| gatctctggcagt ATCTGGGGGCCC ggaggaagtctctttaa | DLWQYLGARRKSL | 1 | |
| gatctctggcagt CCCTCGCCC aagtctctttaagaaaatag | DLWQSLAQVSLRK | 1 | |
| gatctctggcagt CCTTTTCCC aagtctctttaagaaaatag | DLWQSFSQVSLRK | 1 | |
| gatctctggcagt GCTTCGCCA aagtctctttaagaaaatag | DLWQCFAKVSLRK | 1 | |
| gatctctggcagt GTTTTTCAA aagtctctttaagaaaatag | DLWQCFSKVSLRK | 1 | |
| gatctctggcagt TACTTTCCC aagtctctttaagaaaatag | DLWQLLSQVSLRK | 1 | |
| gatctctggcagt TATTTTCCC aagtctctttaagaaaatag | DLWQLFSQVSLRK | 1 | |
| gatctctggcagtgga CCCT ggaagtctctttaagaaaatag | DLWQWTLEVSLRK | 1 | |
| gatctctggcagtgga TGGA ggaagtctctttaagaaaatag | DLWQWMEEVSLRK | 1 | |
| gatTtTtggcagGggaggaagtTtTtttaagaaaatag | DFWQGRKFF | 1 | |
| gatctctggcagGggaggaagCctctttaagaaaatag | DLWQGRKPL | 1 |
| Type | Nucleotide sequence of NPM1 mutations | Translation | Occurrence |
|---|---|---|---|
| WT | gatctctggcagtggaggaagtctctttaa | DLWQWRKSL | NA |
| gatctct ATCT ggcagtggaggaagtctctttaagaaaatag | DLYLAVEEVSLRK | 1 | |
| gatctctg CAGG gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 1 | |
| B | gatctctg CATG gcagtggaggaagtctctttaagaaaatag | DLCMAVEEVSLRK | 18 |
| gatctctg CCGG gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 3 | |
| D | gatctctg CCTG gcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 8 |
| gatctctg CGGA gcagtggaggaagtctctttaagaaaatag | DLCGAVEEVSLRK | 1 | |
| gatctctg CTTG gcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 1 | |
| gatctctg TACG gcagtggaggaagtctctttaagaaaatag | DLCTAVEEVSLRK | 1 | |
| gatctctg TATG gcagtggaggaagtctctttaagaaaatag | DLCMAVEEVSLRK | 1 | |
| gatctctg TCAG gcagtggaggaagtctctttaagaaaatag | DLCQAVEEVSLRK | 1 | |
| gatctctg TCGG gcagtggaggaagtctctttaagaaaatag | DLCRAVEEVSLRK | 1 | |
| A | gatctctg TCTG gcagtggaggaagtctctttaagaaaatag | DLCLAVEEVSLRK | 43 |
| gatctctg TGCC gcagtggaggaagtctctttaagaaaatag | DLCAAVEEVSLRK | 1 | |
| gatctctg TGTG gcagtggaggaagtctctttaagaaaatag | DLCVAVEEVSLRK | 2 | |
| gatctctggc TCCGATTTGC ggaagtctctttaagaaaatag | DLWLRFAEVSLRK | 1 | |
| gatctctggca AGATCTCAGCAAG gtctctttaagaaaatag | DLWQDLSKVSLRK | 1 | |
| gatctctggcag CGGA tggaggaagtctctttaagaaaatag | DLWQRMEEVSLRK | 1 | |
| gatctctggcag CGGATGGCC gaagtctctttaagaaaatag | DLWQRMAEVSLRK | 1 | |
| gatctctggcag CGGATTCC ggaagtctctttaagaaaatag | DLWQRIPEVSLRK | 2 | |
| gatctctggcag CGTTC ggaggaagtctctttaagaaaatag | DLWQRSEEVSLRK | 1 | |
| gatctctggcagt ATCTGGGGGCCC ggaggaagtctctttaa | DLWQYLGARRKSL | 1 | |
| gatctctggcagt CCCTCGCCC aagtctctttaagaaaatag | DLWQSLAQVSLRK | 1 | |
| gatctctggcagt CCTTTTCCC aagtctctttaagaaaatag | DLWQSFSQVSLRK | 1 | |
| gatctctggcagt GCTTCGCCA aagtctctttaagaaaatag | DLWQCFAKVSLRK | 1 | |
| gatctctggcagt GTTTTTCAA aagtctctttaagaaaatag | DLWQCFSKVSLRK | 1 | |
| gatctctggcagt TACTTTCCC aagtctctttaagaaaatag | DLWQLLSQVSLRK | 1 | |
| gatctctggcagt TATTTTCCC aagtctctttaagaaaatag | DLWQLFSQVSLRK | 1 | |
| gatctctggcagtgga CCCT ggaagtctctttaagaaaatag | DLWQWTLEVSLRK | 1 | |
| gatctctggcagtgga TGGA ggaagtctctttaagaaaatag | DLWQWMEEVSLRK | 1 | |
| gatTtTtggcagGggaggaagtTtTtttaagaaaatag | DFWQGRKFF | 1 | |
| gatctctggcagGggaggaagCctctttaagaaaatag | DLWQGRKPL | 1 |
Inserted bases are shown in bold capital letters in the second column. The 4 conserved hydrophobic positions of the nuclear export signal are in bold-face type in the third column. The first base shown is c.856 from the coding reference sequence.
The most common mutation was the duplication type A c.860_863dupTCTG (1793/2423, 74.0%), followed by type B c.863_864insCATG (8.9%) and type D c.863_864insCCTG (6.9%) insertions. Neither type B or D mutations meet the HGVS definition of a duplication, but both show duplicated bases separated by nucleotides of unknown origin. The 4 additional bases in a type B mutation consist of 2 bases of unknown origin (CA), followed by a 2-bp duplication (TG), whereas type D mutations contain 1 base of unknown origin (C), followed by a 3-bp duplication (CTG). We further identified 1 additional exact duplication, c.871_872dupTGGA; 142 non-B/non-D duplications with filler; 51 insertions without any duplicated bases; 51 indels; and 2 multiple clustered point mutations that created an NES in the existing ORF. Anatomically, NPM1 mutations therefore resemble miniature FLT3-ITDs, with similar use of junctional filler nucleotides.
Because NPM1 mutation anatomy was consistent with replication slippage, we sought the germline microhomology anticipated for priming, especially for the highly recurrent type A c.860_863dupTCTG duplication. Critically, there was no G at c.859 or T at c.864 and hence no germline microhomology available to prime type A duplications. Although duplications can occasionally be generated by nonhomologous end-joining,20,21 the latter is unlikely to cause a single highly recurrent mutation. There was no evidence of a secondary structure within the breakpoint region.
We also considered whether germline microhomology was available at any other position within the NPM1 insertion region to prime a 4-bp duplication that could lead to a transforming protein. Such mutations might be predicted at high frequency by a replication-based model. The targeted region of NPM1 is smaller than that of FLT3, with start and end points spread over 16 bp (c.860 to c.876) (Tables 1 and2). The start region was just 12 bp (c860. to c.871), with highly uneven use of positions within this region (Table 3). The products of all 12 hypothetical duplications across the start region were assessed (supplemental Table 3). Six of 12 duplications showed germline microhomology, by definition forming 3 identical pairs. Two of these mutations, c.862_865dupTGGC and c.864_867dupGCAG, break the W288 rule, retaining p.Trp288 with a weak l-xxx-V-xx-V-x-L type NES,13 and are likely benign. The third encodes a product not observed in AML. This suggests no germline microhomology is available to prime relevant slippage within the insertion region. We therefore considered whether TdT provides occult microhomology for priming and the filler nucleotides observed in 627/2423 (25.9%) of NPM1 mutations represented N-nucleotides.
Incidence of use of NPM1 insertion positions
| NPM1 Insertion Site | Adult | Pediatric | Adult + pediatric | P | |||
|---|---|---|---|---|---|---|---|
| NPM1 mutations, n | NPM1 mutations, % | NPM1 mutations, n | NPM1 mutations, % | NPM1 mutations, n | NPM1 mutations, % | Adult vs pediatric | |
| 860_861 | 3 | 0.13 | 0 | 0.00 | 3 | 0.13 | N/T |
| 861_862 | 6 | 0.26 | 0 | 0.00 | 6 | 0.25 | N/T |
| 862_863 | 2 | 0.09 | 1 | 1.01 | 3 | 0.13 | N/T |
| 863_864 | 2267 | 97.63 | 82 | 82.83 | 2349 | 97.03 | <.001 |
| 864_865 | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | N/T |
| 865_866 | 0 | 0.00 | 1 | 1.01 | 1 | 0.04 | N/T |
| 866_867 | 2 | 0.09 | 1 | 1.01 | 3 | 0.1 | N/T |
| 867_868 | 26 | 1.12 | 5 | 5.05 | 31 | 1.28 | .008 |
| 868_869 | 10 | 0.43 | 7 | 7.07 | 17 | 0.70 | <.001 |
| 869_870 | 6 | 0.26 | 0 | 0.00 | 6 | 0.25 | N/T |
| 870_871 | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | N/T |
| 871_872 | 0 | 0.00 | 2 | 2.02 | 2 | 0.08 | N/T |
| n = 2322 | n = 99 | n = 2421 | |||||
| NPM1 Insertion Site | Adult | Pediatric | Adult + pediatric | P | |||
|---|---|---|---|---|---|---|---|
| NPM1 mutations, n | NPM1 mutations, % | NPM1 mutations, n | NPM1 mutations, % | NPM1 mutations, n | NPM1 mutations, % | Adult vs pediatric | |
| 860_861 | 3 | 0.13 | 0 | 0.00 | 3 | 0.13 | N/T |
| 861_862 | 6 | 0.26 | 0 | 0.00 | 6 | 0.25 | N/T |
| 862_863 | 2 | 0.09 | 1 | 1.01 | 3 | 0.13 | N/T |
| 863_864 | 2267 | 97.63 | 82 | 82.83 | 2349 | 97.03 | <.001 |
| 864_865 | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | N/T |
| 865_866 | 0 | 0.00 | 1 | 1.01 | 1 | 0.04 | N/T |
| 866_867 | 2 | 0.09 | 1 | 1.01 | 3 | 0.1 | N/T |
| 867_868 | 26 | 1.12 | 5 | 5.05 | 31 | 1.28 | .008 |
| 868_869 | 10 | 0.43 | 7 | 7.07 | 17 | 0.70 | <.001 |
| 869_870 | 6 | 0.26 | 0 | 0.00 | 6 | 0.25 | N/T |
| 870_871 | 0 | 0.00 | 0 | 0.00 | 0 | 0.00 | N/T |
| 871_872 | 0 | 0.00 | 2 | 2.02 | 2 | 0.08 | N/T |
| n = 2322 | n = 99 | n = 2421 | |||||
A Bonferroni correction of 3 was applied, hence a P < .0167 was considered significant. Mutations at positions c.860_861, c.861_862 and c.862_863 may result from loss of 1 to 3 nt from c.863_864.
N/T, not tested.
Characterization of NPM1 fillers
TdT is biased toward addition of G/C nucleotides, resulting in N-nucleotide G/C content ≥57% in antigen receptors.22,23 The G/C content of 1614 NPM1 filler nucleotides was elevated for both adults (57.1%) and children (59.3%) and across all insertion positions (supplemental Table 3). In contrast, flanking genomic DNA of 50 bp to 2 kb, the NPM1 coding sequence and the human genome have G/C content of 30% to 42%. The G/C content of NPM1 filler nucleotides is therefore consistent with synthesis by TdT. In comparison, junction sequences from BCOR-ITDs (short driver duplications found in various solid tumors that lack TdT) did not reveal equivalent G/C rich insertions (J.B., unpublished data 10 August 2019).
N-regions from antigen receptors have a mean length of 2.8 to 4.2 nt (range, 1-13),22,23 whereas N-regions from nonantigen receptor loci are more variable (BTG1, mean 5.8 nt, range, 1-2124 ; FLT3-ITDs, mean 5.6 nt, range, 1-3618 ). The mean length of 627 NPM1 fillers was 2.6 nt, range, 1-17; only 2/627 fillers exceeded 13 nt in length (supplemental Figure 1). NPM1 fillers increased in length while decreasing in frequency, but also showed evidence of bimodality, with a major peak at 2 nt and a minor peak at 9 nt. The 9-nt peak reflects indels at c.868_869 (9-bp insertion; 5-bp deletion). These longer indels may be required to optimize the NES. Overall, the size and range of NPM1 fillers were consistent with N-regions.
A further hallmark of N-regions is runs of homopurines and homopyrimidines caused by nucleotide stacking in the absence of template.25 We analyzed NPM1 fillers for overrepresentation of purine–purine and pyrimidine–pyrimidine (YY) dinucleotides and underrepresentation of RY and YR dinucleotides. The adult dataset contained 452 dinucleotides from 94 mutations; 7 of 8 purine–purine and YY dinucleotides were overrepresented, 3 significantly, whereas 7 of 8 of the RY and YR dinucleotides were underrepresented, 3 significantly (Table 4). Analyses of pediatric (supplemental Table 4) and combined NPM1 and FLT3-ITD dinucleotide datasets (supplemental Table 5) afforded similar conclusions. These data strongly suggest TdT synthesizes NPM1 fillers.
Dinucleotide analysis of N-nucleotides from adult NPM1 mutations
| Dinucleotide | No. obs | No. exp | Obs/exp ratio | P | Significant at P = .05 |
|---|---|---|---|---|---|
| RR* | |||||
| GG | 24 | 17.01 | 1.41 | .065 | No |
| AA | 19 | 16.66 | 1.14 | .318 | No |
| GA | 25 | 16.84 | 1.48 | .032 | Yes |
| AG | 28 | 16.84 | 1.66 | .006 | Yes |
| YY* | |||||
| CC | 54 | 55.69 | 0.97 | .612 | No |
| TT | 47 | 31.26 | 1.50 | .004 | Yes |
| CT | 42 | 41.73 | 1.01 | .497 | No |
| TC | 44 | 41.73 | 1.05 | .370 | No |
| RY† | |||||
| GC | 30 | 30.78 | 0.97 | .493 | No |
| GT | 12 | 23.06 | 0.52 | .008 | Yes |
| AT | 14 | 22.82 | 0.61 | .034 | Yes |
| AC | 15 | 30.46 | 0.49 | .001 | Yes |
| YR† | |||||
| CG | 32 | 30.78 | 1.04 | .638 | No |
| CA | 28 | 30.46 | 0.92 | .378 | No |
| TG | 19 | 23.06 | 0.82 | .228 | No |
| TA | 19 | 22.82 | 0.83 | .258 | No |
| Dinucleotide | No. obs | No. exp | Obs/exp ratio | P | Significant at P = .05 |
|---|---|---|---|---|---|
| RR* | |||||
| GG | 24 | 17.01 | 1.41 | .065 | No |
| AA | 19 | 16.66 | 1.14 | .318 | No |
| GA | 25 | 16.84 | 1.48 | .032 | Yes |
| AG | 28 | 16.84 | 1.66 | .006 | Yes |
| YY* | |||||
| CC | 54 | 55.69 | 0.97 | .612 | No |
| TT | 47 | 31.26 | 1.50 | .004 | Yes |
| CT | 42 | 41.73 | 1.01 | .497 | No |
| TC | 44 | 41.73 | 1.05 | .370 | No |
| RY† | |||||
| GC | 30 | 30.78 | 0.97 | .493 | No |
| GT | 12 | 23.06 | 0.52 | .008 | Yes |
| AT | 14 | 22.82 | 0.61 | .034 | Yes |
| AC | 15 | 30.46 | 0.49 | .001 | Yes |
| YR† | |||||
| CG | 32 | 30.78 | 1.04 | .638 | No |
| CA | 28 | 30.46 | 0.92 | .378 | No |
| TG | 19 | 23.06 | 0.82 | .228 | No |
| TA | 19 | 22.82 | 0.83 | .258 | No |
Calculated using frequencies of individual nucleotides G = 0.194, A = 0.192, T = 0.263, and C = 0.351.
For the RR and YY dinucleotides, the P value reflects the cumulative binomial probability of the observed value exceeding or equalling the expected value.
For RY and YR dinucleotides, the P value is for observed values less than or equal to the expected value.
Model for NPM1 mutation genesis
The data here suggest a model of replication slippage comparable to FLT3-ITD genesis,18 but without use of germline microhomology. To create a type A duplication, TdT would add a T to c.863, allowing illegitimate alignment with position 860. Polymerization from here leads to the TCTG duplication (Figure 1A). Because the T is used for priming, no N-nucleotides are visible in the final mutation. To create a type D c.863_864insCCTG, TdT adds 2 nucleotides, CC. The second of these bases primes from 861, leaving the first C visible as an N-nucleotide at the center of a 3-bp interrupted duplication (Figure 1B). Type B mutations occur through addition of 3 nucleotides (CAT) at 863_864, with the 3′ T priming from 862, leaving the CA visible as an N-region within a 2-bp interrupted duplication (Figure 1C). To create a 4-bp insertion, TdT adds 5 nt, with the fifth nucleotide used for priming (Figure 1D). Addition of >5 nt by TdT will lead to indel formation through reintegration at positions progressing toward position 877. The longest N-region was a 17-nt TCGGAGTCTCGGCGGAC synthesis associated with a 13-bp deletion.

TdT/occult microhomology model for genesis of NPM1 mutations. (A) Type A mutation, single base of occult microhomology. (B) Type D mutation, 2 nucleotides added, the first visible as an N-nucleotide, the second used for occult microhomology. (C) Type B mutation, 3 nucleotides added, 2 visible as N-nucleotides. (D) c.863_864insCCGG mutation, 5 nucleotides added, 4 visible as N-nucleotides.
TdT/occult microhomology model for genesis of NPM1 mutations. (A) Type A mutation, single base of occult microhomology. (B) Type D mutation, 2 nucleotides added, the first visible as an N-nucleotide, the second used for occult microhomology. (C) Type B mutation, 3 nucleotides added, 2 visible as N-nucleotides. (D) c.863_864insCCGG mutation, 5 nucleotides added, 4 visible as N-nucleotides.
Incidence prediction of 4-bp insertions at c.863_864
This model implicitly predicts the relative incidence of all insertions at a given site, assuming no bias against particular translation products. For example, there are 256 (44) different 4-bp sequences that could be inserted at c.863_864; their relative incidence is predetermined by TdT’s G/C bias, nucleotide stacking, and preference for shorter syntheses. These parameters are known for FLT3-ITDs, where there is no apparent restriction on N-region length or codon usage (supplemental Table 2). Comparison of the predicted and observed incidences for the 256 theoretical c.863_864 4-bp insertion/duplication mutations (n = 2257 adult mutations) (supplemental Table 6) showed a significant correlation (ρ = 0.484, P < .0001; Spearman rank correlation) (Figure 2). These results provide strong support for the TdT/occult microhomology model. Aspects of this model are explored in the following section.
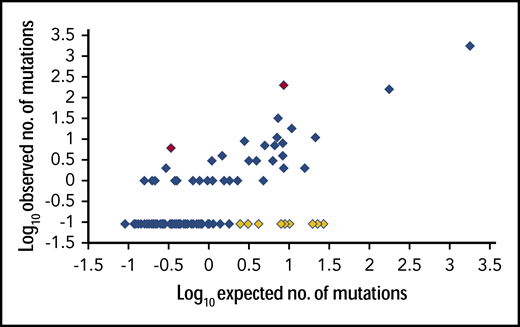
Scatter plot of logarithmic transformations of observed vs predicted numbers of adult NPM1 4-bp insertion/duplication mutations at c.863_864. From a cohort of 2257 independent mutations showing significant correlation between observed and predicted values. Each diamond represents 1 of the 122 possible mutations. Observed counts of 0 were substituted with the predicted occurrence of the rarest mutation to allow logarithmic transformation. Red diamonds: 2 most overrepresented mutations, c.863_844insCATG and c.863_844insCAGA. Yellow diamonds: 10 mutations predicted to occur 2 to 27 times in the cohort, but not observed.
Scatter plot of logarithmic transformations of observed vs predicted numbers of adult NPM1 4-bp insertion/duplication mutations at c.863_864. From a cohort of 2257 independent mutations showing significant correlation between observed and predicted values. Each diamond represents 1 of the 122 possible mutations. Observed counts of 0 were substituted with the predicted occurrence of the rarest mutation to allow logarithmic transformation. Red diamonds: 2 most overrepresented mutations, c.863_844insCATG and c.863_844insCAGA. Yellow diamonds: 10 mutations predicted to occur 2 to 27 times in the cohort, but not observed.
The 4-bp insertions at c.863_864 encode the last base of codon 288 and the entire new codon 289; germline W288 (tryptophan) is encoded by TGG, hence synthesis of N-nucleotides initiates after the TG (Figure 1A). FLT3-ITD data suggest TdT adds just a single nucleotide 54.2% of the time. Creation of a functional NPM1 mutation is a molecular roulette. The probabilities of TdT adding a G, A, T, or C following a G are 0.58, 0.21, 0.09, and 0.12, respectively (values from FLT3-ITD N-regions; supplemental Table 2). If chance favors G, priming cannot occur from the required position to create a 4-bp insertion. The lesion might be repaired by reintegration elsewhere, but is unlikely to promote AML. Should TdT add additional nucleotide(s) that do permit priming of a comparable 4-bp frameshift, then the original codon 288 would be recreated to encode tryptophan; the resulting NES would break the W288 rule and fail to transform. None of the 64 4-bp insertions starting with a G were observed. Alternatively, if TdT adds an A as the first nucleotide after c.863, followed by further nucleotide(s) to allow reintegration in the appropriate frame, a TGA stop codon is created. The absence of all 64 4-bp insertions starting with A shows truncated NPM1 is not transforming. The pool of potential mutations is therefore reduced to 128. In contrast, although addition of a C alone does not permit correct priming for c.863_864ins(4) mutations, chain elongation will permit integration that can create a leukemogenic mutant.
However, if a T is added after c.863, a type A mutation can be formed through mispriming from nucleotide 860, as shown (Figure 1A). The likelihood of this is obtained by multiplying the frequency of obtaining a single base TdT extension (0.542) by the incidence of a G to T extension by TdT (0.09; supplemental Table 2; derived from FLT3 N-region data). To obtain the expected incidence, this value must be corrected to reflect events that do not result in a functional protein, either because they result in a stop or lack the correct occult microhomology for priming in the required frame; only 6.7% of all 1 through 5 nucleotide extensions from c.863 yield a potentially functional protein. We must further allow for the additional ways this mutation can occur: 2 through 5 nucleotide extensions of TC, TCT, TCTG, and TCTGG. Summing these probabilities shows the expected incidence of a type A mutation is 79.6%. Among 2257 mutations, we would therefore expect 1797 type A mutations; 1750 were observed.
As expected, only mutations starting with T or C were observed. The number of possible mutations can be further reduced by 6 to reflect the 3 stop codons preceded by T or C; the total number of mutations considered possible was therefore 122. The rarest predicted mutations were c.863_844insCGCT and c.863_844insCTGC (expected incidence <0.001%) (supplemental Table 6). To create these mutations, TdT must add 5 N-nucleotides, with the fifth nucleotide, G, providing occult microhomology; such extensions provide many permutations. The incidence of c.863_844insCGCT is the product of the relative incidences of GC (0.12), CG (0.17), GC (0.12), CT (0.08), and TG (0.28) extensions, corrected for the incidence of 5 nucleotide extensions (5.08%) and for unsuccessful events. These mutations require purine to pyrimidine switches (or vice versa) in 4/5 of the polymerizations (the maximum number of switches possible when starting and ending on a purine). Such switches are not favored by TdT because of its preference to stack nucleotides. The incidence of individual NPM1 mutation 4-bp duplication/insertions at c.863 therefore varies over at least 4 orders of magnitude.
The second most common extension length is 2 nt. Only 2 such extensions are viable, TC and CC, with the second base providing occult microhomology. TC yields a type A mutation, CC a type D c.863_864insCCTG. A total of 177 type D mutations were predicted; 160 were observed. There are 8 3-nt extension length mutations, all predicted at frequencies that should permit their detection here. All were observed, including the types A and D already discussed (TGTG: expected [exp] 9, observed [obs] 2; TATG: exp 11, obs 18; TTTG: exp 16, obs 2; TCTG: type A; CGTG: exp 6, obs 3; CATG: exp 9, obs 197; CTTG: exp 7, obs 32; and CCTG: type D; supplemental Table 6). The exception among these concordant results was the CATG type B mutation, which was observed more frequently than expected (Figure 2). Other mutations with a CA root were not overrepresented, with the exception of CAGA. It is unclear why some CA root mutations are favored.
The model also predicts the incidence of individual mutations will fall as the number of nucleotide additions by TdT increases, partly because longer extensions are rarer but mainly because the overall number of potential mutations is increased with each extension. However, mutations with longer N-regions can occur more frequently than those with shorter N-regions if they are G/C rich and show significant nucleotide stacking. For example, c.863_864insCCGG occurs more frequently than 3 of the 2-nt N-region mutations.
The calculations behind the model assume codon 289, coded entirely by the insertion, can accommodate any amino acid. Codon 289 represents the third amino acid in the L-xxx-V-xx-V-x-L NES, and a wide range of amino acids is clearly tolerated at this position (Tables 1 and 2). However, intolerance of some amino acids is suggested by the absence of certain predicted mutations (supplemental Table 6; Figure 2). Strikingly, none of the 8 4-bp insertions encoding p.Pro289 were observed (combined predicted incidence 49) (supplemental Tables 7 and 8), suggesting intolerance of p.Pro289. Activity-based profiles of NESs predict reduced NES activity when proline is incorporated at position 3.26 Our results suggest this reduced NES activity may impair creation of a functional NPM1 mutation. Similarly, the 8 mutations encoding p.Gly289 were not observed (supplemental Table 7), although a single mutation encoding p.Gly289 was present in the pediatric cohort (Table 2). Glycine may therefore be poorly tolerated through creation of a weaker (but still functional) NES. Overall, a significant correlation was observed between the predicted and observed amino acid frequencies, even with inclusion of proline and glycine (ρ = 0.512, P = .021; Spearman rank correlation).
Insertions at c.867_868 and c.868_869
The second most common insertion/indel position is c.867_868, although still rare compared with c.863_864 (1.28% vs 97.0%, Table 3). However, the number of mutations observed does not necessarily reflect the level of slippage/targeting of a particular site because only a fraction of mutations may be transforming and this fraction will vary from site to site. We applied the tenets of the TdT model to compare the levels of mutagenesis at these sites.
The c.867_868 mutations consist of 4-bp insertions and longer indels (Tables 1 and2). From the unrestrained FLT3-ITD data, we know that a majority (74%) of TdT extensions consist of 1 to 4 nucleotides, leading to N-regions of 1 to 3 nucleotides. However, all 4-nt insertions/duplications at c.867_868 resulting from addition of 1 to 4 nucleotides to c.867G are predicted to lead to duplication of G in the 4th and final position of the insertion. This G, with the following TG, creates a GTG codon encoding valine at p.291, thereby breaking the W288 rule (the requirement to create a stronger NES when W288 is retained). The majority of slippage events at c.867_868 will therefore not be observed.
However, if TdT adds ≥5 nucleotides to c.867G, then alternatives to p.Val291 become possible. The fifth nucleotide is used for occult microhomology, and these mutations therefore appear as 4-bp insertions. There were 20 adult 4-bp pure insertions at c.867_868, all with C, A, or T as the final nucleotide (Table 1). In comparison, there were 22 pure insertions at c.863_864 (Table 5). This suggests at least equivalent levels of slippage at the 2 sites, whereas constraints on amino acid usage at p.290 (encoded entirely by the N-region) in c.867_868ins(4) mutations suggests slippage at c.867_868 may be higher than at c.863_864.
NPM1 c.863_864 4 bp insertion/duplication mutations, according to their number of N-nucleotides
| No. N-nucleotides added | No. adult mutations observed (n = 2257) | Adult: % observed | Expected in population of n = 82 | No. of pediatric mutations observed (n = 82) | Pediatric: ratio predicted/observed | P |
|---|---|---|---|---|---|---|
| 1 | 1750 | 77.5 | 63.6 | 43 | 0.7 | <.001 |
| 2 | 160 | 7.1 | 5.8 | 8 | 1.4 | .379 |
| 3 | 254 | 11.3 | 9.2 | 22 | 2.4 | <.001 |
| 4 | 71 | 3.1 | 2.6 | 7 | 2.7 | .018 |
| 5 | 22 | 1.0 | 0.8 | 2 | 2.5 | .205 |
| 3-5 | 347 | 15.4 | 12.6 | 31 | 2.5 | <.001 |
| No. N-nucleotides added | No. adult mutations observed (n = 2257) | Adult: % observed | Expected in population of n = 82 | No. of pediatric mutations observed (n = 82) | Pediatric: ratio predicted/observed | P |
|---|---|---|---|---|---|---|
| 1 | 1750 | 77.5 | 63.6 | 43 | 0.7 | <.001 |
| 2 | 160 | 7.1 | 5.8 | 8 | 1.4 | .379 |
| 3 | 254 | 11.3 | 9.2 | 22 | 2.4 | <.001 |
| 4 | 71 | 3.1 | 2.6 | 7 | 2.7 | .018 |
| 5 | 22 | 1.0 | 0.8 | 2 | 2.5 | .205 |
| 3-5 | 347 | 15.4 | 12.6 | 31 | 2.5 | <.001 |
A Bonferroni correction of 6 was applied, hence P < .0083 was considered significant.
Pediatric vs adult mutations
NPM1 mutations from children have a lower proportion of type A mutations than adults.5,6,16,17 A key difference between type A and non-type A mutations is that type A mutations only require a single nucleotide addition by TdT, whereas the latter require 2 or more. The simplest explanation to account for the age-specific difference is therefore a higher level of TdT activity in pediatric myeloid stem cells. Increased TdT activity would bypass type A mutations in favor of those with longer extensions. We confirmed a significantly lower incidence of type A mutations in children (43/101 [42.6%], cf 1750/2322 [75.4%] in adults; P < .001, Fisher’s exact test), then tested the predictions imposed by this solution.
First, N-regions should be longer on average in children than adults. Restricting analysis to mutations with visible N-nucleotides, the mean N-region in children was 3.80 nt (n = 55), cf 2.46 nt in adults (n = 572). Repeating the analysis with inclusion of all nonpoint mutations confirmed this finding; pediatric mean 2.11 nt (n = 99), adult mean 0.61 nt (n = 2322). However, these figures exclude the additional N-nucleotide required for occult microhomology. The mean numbers of total nucleotides added in children and adults are therefore 3.11 and 1.61, respectively, consistent with twofold higher TdT activity at younger age.
Second, this solution predicts differential use of insertion sites in adults and children. Insertions at c.867_868 and c.868_869 are longer than those at c.863_864 in both adults and children (Tables 1 and2). As discussed, replication errors at these alternative sites are predicted to occur more commonly than suggested by their observed incidence. With increased TdT activity, more NPM1 insertions should reach the required length to use these alternative sites. Both of these sites are used at significantly increased frequency in children (ie, for c.867_868 insertions, P = .008, Fisher’s exact test) (Table 3).
Third, the model implies a different spectrum of c.863_864ins(4) mutations in children. Although type A mutations occur at decreased frequency, changes in the frequency of the other c.863_864ins(4) mutations should vary according to the number of TdT additions (2-5 nt) required for their genesis; the incidence of type D and type B mutations, requiring addition of 2 and 3 nucleotides, respectively, may not change equally. We categorized all c.863_864ins(4) mutations by number of TdT nucleotide additions and compared the results between age groups (Table 5). Mutations requiring a single nucleotide are significantly underrepresented in children (P ≤ .001, Fisher’s exact test); those requiring 3 to 5 nucleotides are significantly overrepresented (31/83 [37.8%] in children, cf 347/2257 [15.4%] in adults; P ≤ .001 Fisher’s exact test), whereas the incidence of 2 nucleotide extension mutations was not significantly changed (Table 5). In summary, the childhood incidences of type A, D, and B mutations are decreased, unchanged, and increased respectively. These results support the idea of differential TdT expression between adults and children, and the wider model that TdT drives NPM1 mutagenesis.
Discussion
In our accompanying paper, we suggest TdT plays a significant role in AML by priming replication slippage to generate FLT3-ITDs. Here we propose that NPM1 mutations also arise following illegitimate TdT activity. Analysis of 2430 NPM1 mutations confirms and extends the FLT3-ITD findings in an independent dataset using shared and complementary approaches. These findings propel TdT to membership of a select group of mutagenic lymphoid enzymes normally harnessed to promote antigenic diversity, but whose illegitimate activities promote cancer. These enzymes include RAG, which physiologically mediates V(D)J recombination at antigen receptor loci, but can cause oncogenic translocations in acute lymphoblastic leukemia and lymphoma27 ; and AID, a deaminating enzyme required for somatic hypermutation and class switch recombination that also induces off-target damage.28 RAG, AID, and TdT therefore promote hematological neoplasia through distinct mutagenic activities. TdT is predicted to both prevent correct reintegration and promote mispriming during slippage.
We recognize that TdT is not expressed at high levels in NPM1-mutated AML at presentation and suggest that TdT levels may be down-regulated subsequent to mutation acquisition. There is a parallel for this in the presence of IG/TCR gene rearrangements in AML blasts that no longer retain expression of the RAG1/2 genes.29
The shared origin of FLT3-ITD and NPM1 mutations explains their similar molecular anatomy. For both types of mutation, addition of nucleotides in excess of that required for priming results in visible G/C-rich N-regions, with significant nucleotide stacking, located between the 2 copies of the repeat. However, unlike FLT3-ITDs, NPM1 mutations invariably require priming by TdT, reflecting a lack of appropriately positioned germline microhomology. The marked cooccurrence between NPM1 and FLT3-ITD mutations in AML2,5,6 may in part be explained through this common origin, although these mutations are also recognized to show significant molecular synergy.19
The chance of creating a transforming NPM1 mutation is defined through the dictates of a molecular roulette following addition of nucleotide(s) by TdT. There are only limited positions at which mispriming can occur that will lead to a transforming frameshift mutation, and such priming requires addition of the requisite complementary nucleotide. Choice of nucleotide is biased by TdT’s G/C and stacking preferences, but each addition remains a chance event. Highly recurrent NPM1 mutations reflect early addition of the correct N-nucleotide for appropriate priming. As the number of nucleotides added by TdT increases, the number of sequence permutations and possible mutations increases, and each mutation occurs at decreased frequency. The successful prediction of the incidence of the 4-bp 863_864 NPM1 insertions helps verify the TdT-mutator model. The success rate for creating functional NPM1 mutations may be lower than for FLT3-ITDs because, although one-third of FLT3 reintegrations will occur in frame through integration across a ∼100-bp region, NPM1 mutations may only have a single appropriate site. Elsewhere in AML genomes, indels occur only infrequently for reasons that remain unclear, but might reflect in part a need for a secondary structure (such as the FLT3 palindrome) to facilitate access by TdT.
We also suggest why the NPM1 mutation spectrum differs with age: a higher level of TdT activity in children may cause a shift away from type A mutations toward mutations with increasing number of N-nucleotides, including those at less commonly used insertion sites. TdT activity is known to vary with age.30-32 This explains some of the genetic differences between adult and pediatric AML.33
In summary, the implication of TdT as a mutagen in AML is a significant finding, with key roles proposed in both FLT3-ITD and NPM1 mutagenesis. Even allowing for the concurrence between these mutations, around one-half of all AML cases may arise following off-target TdT activity. Other AML mutations, such as KIT-ITDs, may also involve TdT. An increased incidence of AML may therefore be part of the price we pay for adaptive immunity.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgment
The authors thank Joanne Mason (West Midlands Regional Genetics Laboratory) for critical reading of the manuscript.
Authorship
Contribution: J.B. conceived the study, analyzed data, performed statistical analyses, and wrote the draft manuscript; S.A.D., S.A., and M.J.G. supervised the project; and all authors provided intellectual input, revised, and gave final approval to the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Julian Borrow, West Midlands Regional Genetics Laboratory, Birmingham Women’s and Children’s NHS Foundation Trust, Mindelsohn Way, Edgbaston, Birmingham, B15 2TG, United Kingdom; e-mail: j.borrow@nhs.net.
