

TO THE EDITOR:
One of the most common reasons for hematology consultation is the evaluation of cytopenia(s). The workup of patients who present with cytopenia(s) can be extensive, given the wide differential diagnosis.1 Although only a minority of patients are ultimately diagnosed with a hematologic malignancy, a key entity to exclude in this differential is myelodysplastic syndrome (MDS),2 which requires bone marrow morphology and cytogenetics for diagnosis. There is a clinical need for the development of minimally invasive ancillary tests to enhance conventional hematologic workup (eg, complete blood count with differential, B12/folate testing, iron-related studies, and serum protein electrophoresis) in the identification of patients who are at a low risk of having an underlying hematologic malignancy as the cause of cytopenia(s), thereby avoiding a costly and invasive bone marrow biopsy (BMBx).
Mutation profiling of peripheral blood (PB) using next-generation sequencing (NGS) is an attractive solution to this problem because of its potential application as a minimally invasive screen. Recent large-scale genome-sequencing studies using bone marrow samples have demonstrated that most cases of MDS and other related neoplasms, such as acute myeloid leukemia and myelodysplastic/myeloproliferative neoplasm overlap syndromes, harbor pathogenic somatic mutations in diverse myeloid cancer driver genes.2-6 Moreover, some or all of these mutations can be detected in PB granulocytes in most patients with MDS and related neoplasms (Phillip D. Michaels, Dahai Wang, A.S.K., manuscript in preparation).7,8
Despite these advances and the widespread use of NGS testing, there are limited data on the clinical use of NGS testing in the early evaluation of patients with cytopenia(s). We hypothesize that targeted PB NGS using a myeloid cancer gene panel can be a valuable minimally invasive, ancillary tool in identifying patients with an underlying myeloid neoplasm as the cause of cytopenia(s). Herein, we report the clinical utility of PB screening by targeted NGS testing in a large institutional cohort of patients with cytopenia(s).
After institutional review board approval, we retrospectively identified all patients presenting with PB cytopenia(s) over a 30-month period (January 2015 through June 2017) to the Hematology Clinic at the Dana-Farber/Brigham and Women’s Cancer Center. Patients with a known history of a hematologic malignancy were excluded. All patients received a conventional hematologic workup to determine the cause of the cytopenia(s). NGS testing was performed at the discretion of the hematologist on PB of a subset of patients, using a custom 95-gene, amplicon-based sequencing panel surveying genes recurrently mutated in hematologic (myeloid and selected leukemic lymphoid) malignancies.9 A subset of patients also underwent a concurrent BMBx, defined as within 6 months of initial evaluation with no overt clinical change in the interim. The patients were observed through the end of the study period (1 August 2018) or the date of last follow-up, and any subsequent bone marrow biopsies or diagnoses of hematologic malignancies were noted. Statistical analysis was performed with R and GraphPad Prism 7.0.
A total of 1586 patients presenting with cytopenia(s) were identified (Figure 1A). Of these, 276 patients (Table 1) without preexisting history of a hematologic malignancy underwent NGS testing of a PB specimen in the context of a conventional hematologic evaluation. Overall, 77 (28%) patients had a pathogenic mutation in at least 1 of the 95 genes evaluated (Figure 1A; supplemental Figure 2, available on the Blood Web site). Patients in this group were older than those without a pathogenic mutation (median age, 71 vs 61 years; P < .0001), had a higher rate of concurrent bone marrow biopsy (43% vs 20%; P = .0002), and were more likely to present with anemia (86% vs 59%; P = .0048) and lower hemoglobin levels (median 11.2 vs 12.3 g/dL; P = .006). Patients without a pathogenic mutation were more likely to present with a single cytopenia (65% vs 51%; P = .028), whereas patients with a pathogenic mutation were more likely to present with cytopenias of all 3 lineages (19% vs 8.5%; P = .019). However, the 2 groups were comparable in terms of sex and degree of cytopenia(s). The group with pathogenic mutations had a much higher risk of being concurrently diagnosed with a hematologic malignancy when compared with the group without pathogenic mutations (Figure 1A; 27 of 77, 35%, vs 13 of 199, 6.5%; P < .0001). This difference in risk was particularly striking for myeloid neoplasms (19 of 77, 25%, vs 2 of 199, 1%; P < .0001). The 19 myeloid neoplasms diagnosed in patients with a pathogenic mutation included MDS (n = 11), myelodysplastic/myeloproliferative neoplasm overlap (n = 4), therapy-related myeloid neoplasms (n = 3), and systemic mastocytosis with an associated hematological neoplasm (n = 1; supplemental Table 1). In patients without a pathogenic mutation, only 2 patients were diagnosed with MDS (supplemental Table 2). These results suggest that, after a conventional hematologic workup, the absence of a pathogenic mutation identifies patients at very low risk of an underlying (concurrent) myeloid neoplasm as the cause of cytopenia(s).
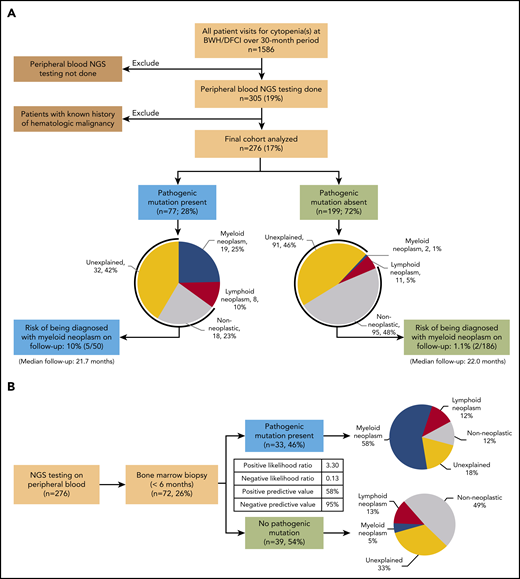
Flow diagrams showing patient selection and results of PB NGS testing and biopsy diagnosis. (A) Patient selection, results of PB NGS testing, diagnosis after hematologic workup, and rate of progression to frank myeloid neoplasm on follow-up. (B) Results of PB NGS testing and short-interval bone marrow biopsy diagnosis (n = 72). The inset table shows PPVs and NPVs and likelihood ratios.
Flow diagrams showing patient selection and results of PB NGS testing and biopsy diagnosis. (A) Patient selection, results of PB NGS testing, diagnosis after hematologic workup, and rate of progression to frank myeloid neoplasm on follow-up. (B) Results of PB NGS testing and short-interval bone marrow biopsy diagnosis (n = 72). The inset table shows PPVs and NPVs and likelihood ratios.
Characteristics of patients who underwent PB NGS testing and a hematologic work-up for cytopenia(s) (n = 276)
| Entire cohort | Pathogenic mutation present | Pathogenic mutation absent | P | |
|---|---|---|---|---|
| Patients, n | 276 | 77 | 199 | |
| Sex, n (M/F) | 135/141 | 40/37 | 95/104 | .59 |
| Age, median y (IQR) | 66 (53-73) | 71 (67-79) | 61 (49-71) | <.0001 |
| Hemoglobin, median g/dL (IQR) | 11.8 (10-13.2) | 11.2 (9.5-12.5) | 12.3 (10.3-13.3) | .006 |
| White blood cells, median K/µL (IQR) | 4.74 (3.34-6.54) | 4.91 (3.59-7.2) | 4.72 (3.29-6.3) | .061 |
| Absolute neutrophil count, median K/µL (IQR) | 2.78 (1.74-4.23) | 2.9 (1.65-4.38) | 2.76 (1.79-4.17) | .17 |
| Platelets, median K/µL (IQR) | 149 (98-224) | 141 (88-217) | 155 (99-226) | .85 |
| Abnormal CBC finding at initial visit, n (%) | ||||
| Anemia | 178 (64) | 60 (86) | 118 (59) | .0048 |
| Leukopenia | 106 (38) | 29 (38) | 77 (39) | .89 |
| Thrombocytopenia | 131 (47) | 41 (53) | 90 (45) | .28 |
| Number of cytopenias, n (%) | ||||
| 1 | 169 (61) | 39 (51) | 130 (65) | .028 |
| 2 | 75 (27) | 23 (30) | 52 (26) | .55 |
| 3 | 32 (12) | 15 (19) | 17 (8.5) | .019 |
| Cytopenia(s) defined as absolute neutrophil count <1.8 × 103/µL, hemoglobin <10.0 g/dL, platelet count < 100 × 103/µL, n (%) | 163 (59) | 52 (67) | 111 (56) | .48 |
| Bone marrow biopsy at initial evaluation, n (%) | 72 (26) | 33 (43) | 39 (20) | .0002 |
| Entire cohort | Pathogenic mutation present | Pathogenic mutation absent | P | |
|---|---|---|---|---|
| Patients, n | 276 | 77 | 199 | |
| Sex, n (M/F) | 135/141 | 40/37 | 95/104 | .59 |
| Age, median y (IQR) | 66 (53-73) | 71 (67-79) | 61 (49-71) | <.0001 |
| Hemoglobin, median g/dL (IQR) | 11.8 (10-13.2) | 11.2 (9.5-12.5) | 12.3 (10.3-13.3) | .006 |
| White blood cells, median K/µL (IQR) | 4.74 (3.34-6.54) | 4.91 (3.59-7.2) | 4.72 (3.29-6.3) | .061 |
| Absolute neutrophil count, median K/µL (IQR) | 2.78 (1.74-4.23) | 2.9 (1.65-4.38) | 2.76 (1.79-4.17) | .17 |
| Platelets, median K/µL (IQR) | 149 (98-224) | 141 (88-217) | 155 (99-226) | .85 |
| Abnormal CBC finding at initial visit, n (%) | ||||
| Anemia | 178 (64) | 60 (86) | 118 (59) | .0048 |
| Leukopenia | 106 (38) | 29 (38) | 77 (39) | .89 |
| Thrombocytopenia | 131 (47) | 41 (53) | 90 (45) | .28 |
| Number of cytopenias, n (%) | ||||
| 1 | 169 (61) | 39 (51) | 130 (65) | .028 |
| 2 | 75 (27) | 23 (30) | 52 (26) | .55 |
| 3 | 32 (12) | 15 (19) | 17 (8.5) | .019 |
| Cytopenia(s) defined as absolute neutrophil count <1.8 × 103/µL, hemoglobin <10.0 g/dL, platelet count < 100 × 103/µL, n (%) | 163 (59) | 52 (67) | 111 (56) | .48 |
| Bone marrow biopsy at initial evaluation, n (%) | 72 (26) | 33 (43) | 39 (20) | .0002 |
Data are shown for entire cohort and are also stratified by the presence or absence of a pathogenic mutation in PB. P values were calculated using the unpaired Student t test (with or without Welch’s correction) for continuous variables or Fisher’s exact test for nominal variables. Bold indicates statistical significance. Cytopenia(s) was defined as absolute neutrophil count, <1.8 × 103/µL; hemoglobin, <10.0 g/dL; and platelet count, < 100 × 103/µL.11 Time frame for bone marrow biopsy at initial evaluation was <6 mo with no clinical intervention.
CBC, complete blood count; IQR, interquartile range.
To investigate further the diagnostic utility of PB mutational profiling as a screening test in predicting the presence of a myeloid neoplasm on BMBx and to control for differences in BMBx rates, we restricted our analysis to patients who underwent a BMBx within 6 months of PB NGS (n = 72; Figure 1B). The absence of a pathogenic mutation in PB was highly predictive of the absence of a concurrent myeloid neoplasm on BMBx (negative predictive value [NPV], 95%; 95% CI, 83%-99%; negative likelihood ratio, 0.13; 95% CI, 0.03-0.50). Conversely, the presence of a pathogenic mutation modestly predicted the presence of a myeloid neoplasm (positive predictive value [PPV]: 58%; 95% CI, 46%-68%; positive likelihood ratio: 3.30; 95% CI, 2.1-5.3). Identical PPV and NPV could also be achieved with a smaller panel of only 20 genes and similar PPV and NPV (93% NPV and 60% PPV) with a panel of as few as 10 genes (supplemental Table 3). The presence of multiple pathogenic variants (2 or more) and larger clone sizes (>20% variant allele fraction) had higher positive predictive power (PPV: 75%; 95% CI, 56%-88% and 79%; 95% CI, 62%-88%, respectively; supplemental Table 4; supplemental Figure 1), similar to findings in a previous study.10 In particular, clone size was a powerful independent predictor of the presence of a concurrent myeloid neoplasm in univariate and multivariate analyses, with sensitivity of 90.5% and specificity of 94.1% (supplemental Tables 4 and 5; supplemental Figure 1; P < .0001). Mutations in the spliceosome pathway were the most predictive of a concurrent myeloid neoplasm (PPV, 79%) but were not significant in multivariate analysis (supplemental Tables 5 and 6).
We also examined the risk of being diagnosed with a myeloid neoplasm on follow-up among patients without a diagnosis of hematologic malignancy at initial evaluation. Among the 199 (72%) patients without a pathogenic mutation, 95 (48%) had a nonneoplastic etiology of cytopenia(s), and 91 (46%) had unexplained cytopenia(s), including 13 with BMBx-confirmed idiopathic cytopenia of uncertain significance. With a median follow-up of 22.0 months, only 2 patients (2 of 186; 1.1%) went on to be diagnosed with a myeloid neoplasm (both MDS; supplemental Table 7). Both of these patients continued to demonstrate an absence of pathogenic mutations in PB or bone marrow in repeat NGS testing. Therefore, after a conventional hematologic workup and lack of a pathogenic mutation, the risk of developing or being diagnosed with a myeloid neoplasm on follow-up is low.
Moreover, among the 77 patients with a pathogenic mutation, 18 (23%) had a clinically diagnosed nonneoplastic etiology of cytopenia(s), and 32 (42%) had unexplained cytopenia(s), including 6 patients with BMBx-confirmed clonal cytopenia of uncertain significance. During a median follow-up of 21.7 months, 5 patients (5 of 50; 10%) with a myeloid mutation and clinical diagnosis of nonmalignant etiology or unexplained cytopenia(s) on initial evaluation were diagnosed with a myeloid neoplasm (supplemental Table 7). Given that they did not uniformly undergo a bone marrow biopsy at initial evaluation, it is not possible to determine at what point in time the disease developed.
In conclusion, we demonstrated that, in an institutional cohort of patients presenting with cytopenia(s) and deemed to be potentially at risk of an underlying hematological malignancy after a conventional hematology workup, routine targeted NGS testing is a valuable ancillary tool for predicting patients at low risk of having a concurrent or future myeloid neoplasm. These findings indicate that PB NGS screening can preselect patients with cytopenia(s) who are unlikely to have an underlying myeloid neoplasm and thereby reduce the need for costly and invasive BMBx testing.
For related data, please send an e-mail to the corresponding author.
The online version of this article contains a data supplement.
Acknowledgment
The authors thank Nancy Berliner for thoughtful comments and suggestions during manuscript preparation.
Authorship
Contribution: A.S.K. and E.A.M conceived of the project; V.S., A.S.K., E.A.M., and A.P. designed the study; V.S., R.K., A.S.K., E.A.M., and A.P. performed the data collection; V.S. performed the data analyses; and V.S., A.S.K., E.A.M., and A.P. wrote the manuscript.
Conflict-of-interest disclosure: During this study, A.S.K. received consulting fees from LabCorp, Inc., Papgene, Inc., and Aushon/Quanterix, Inc. The remaining authors declare no competing financial interests.
Correspondence: Annette S. Kim, Department of Pathology, Harvard Medical School, Brigham and Women’s Hospital, 75 Francis St, Boston, MA 02115; e-mail: askim@bwh.harvard.edu; and Elizabeth A. Morgan, Department of Pathology, Harvard Medical School, Brigham and Women’s Hospital, 75 Francis St, Boston, MA 02115; e-mail: eamorgan@bwh.harvard.edu.
