

Key Points
OPN localizes to the lung and is critically required for induction of murine TRALI via stimulation of pulmonary PMN accumulation.
The OPN-mediated murine TRALI response is dependent on macrophages and OPN polymerization and is independent from CD44, MIP-2, and IL-6.

Abstract
Transfusion-related acute lung injury (TRALI) is one of the leading causes of transfusion-related fatalities and is characterized by the onset of acute respiratory distress within 6 hours upon blood transfusion. Specific therapies are unavailable. Preexisting inflammation is a risk factor for TRALI and neutrophils (polymorphonuclear neutrophils [PMNs]) are considered to be the major pathogenic cells. Osteopontin (OPN) is a multifunctional protein expressed at sites of inflammation and, for example, is involved in pulmonary disorders, can regulate cellular migration, and can function as a PMN chemoattractant. We investigated whether OPN is involved in TRALI induction by promoting PMN recruitment to the lungs. Using a previously established murine TRALI model, we found that in contrast to wild-type (WT) mice, OPN knockout (KO) mice were resistant to antibody-mediated PMN-dependent TRALI induction. Administration of purified OPN to the OPN KO mice, however, restored the TRALI response and pulmonary PMN accumulation. Alternatively, blockade of OPN in WT mice using an anti-OPN antibody prevented the onset of TRALI induction. Using pulmonary immunohistochemistry, OPN could be specifically detected in the lungs of mice that suffered from TRALI. The OPN-mediated TRALI response seemed dependent on macrophages, likely the cellular source of OPN and OPN polymerization, and independent from the OPN receptor CD44, interleukin 6 (IL-6), and other PMN chemoattractants including macrophage inflammatory protein-2 (MIP-2). These data indicate that OPN is critically required for induction of antibody-mediated murine TRALI through localization to the lungs and stimulation of pulmonary PMN recruitment. This suggests that anti-OPN antibody therapy may be a potential therapeutic strategy to explore in TRALI patients.
Introduction
Transfusion-related acute lung injury (TRALI) is a life-threatening syndrome that occurs within 6 hours of a blood transfusion and is characterized by the onset of acute respiratory distress and the development of noncardiogenic pulmonary edema.1 TRALI is one of the leading causes of transfusion-related fatalities and specific therapies are not available.1,2 The pathophysiology of TRALI is complex and a better understanding is highly warranted.1 Polymorphonuclear neutrophils [PMNs] have been recognized as the major pathogenic cells in the pathophysiology of TRALI1-3 whereas CD4+CD25+FoxP3+ T-regulatory cells and dendritic cells are major protective cells against TRALI induction.1,4 TRALI pathology can be viewed as a 2-hit model,1,5,6 where the first hit represents the underlying clinical condition of the patient (eg, inflammation) and the second hit is conveyed by the transfusion product containing pathogenic antileukocyte antibodies1,7 and/or biological response modifiers (eg, lipids).1,8 The nature of the inflammation in TRALI patients is evident from increased levels of the proinflammatory cytokines interleukin-6 (IL-6) and IL-89,10 as well as the acute-phase protein C-reactive protein,11 which is a diagnostic marker for acute infections and inflammation.12 C-reactive protein was also shown to functionally enable antibody-mediated murine TRALI via synergistically enhancing pulmonary PMN recruitment together with the TRALI-inducing antibodies.13
Osteopontin (OPN) is a matricellular protein with multiple biological functions.14 These functions include both normal physiological processes (eg, regulating bone tissue biomineralization and restricting the growth of calcium crystals in epithelial tissues)15 as well as pathological processes in various disease states such as cancer, atherosclerosis, glomerulonephritis, and several chronic inflammatory diseases.16 OPN can function as an important proinflammatory cytokine and is able to regulate cellular adhesion and migration of various immune cells including PMNs.16 Interestingly, OPN has also been shown to be abundantly expressed in multiple lung diseases, regulating aspects of pulmonary granuloma formation, lung fibrosis, and lung carcinoma.17 Additionally, OPN is upregulated at sites of inflammation and tissue remodeling.18,19
As the pathophysiology of TRALI involves inflammation and pulmonary PMN migration, we hypothesized that OPN may play a role in the onset of TRALI as a pulmonary PMN chemoattractant. We therefore investigated the involvement of OPN in TRALI by using our previously established murine TRALI model, which is based on low-dose lipopolysaccharide (LPS) priming (when mice are housed in a specific pathogen-free [SPF] setting) and CD4+ T-cell depletion, followed by injection of anti–major histocompatibility complex (MHC) class I antibodies.4,20 The results suggest that OPN is critically involved in the development of antibody-mediated TRALI by attracting PMNs toward the lungs and that blocking OPN using anti-OPN antibodies may be a promising potential therapeutic intervention for TRALI.
Methods
Mice
C57BL/6 (H-2b, C57BL/6NCrl) wild-type (WT) mice were obtained from Charles River Laboratories (Sulzfeld, Germany) and C57BL/6 OPN knockout (KO) mice were bred and genotyped at Lund University (Lund, Sweden). All mice were males, 8 to 10 weeks of age, and were housed under SPF conditions for at least 1 week in the animal facility at the Biomedical Service Division at Lund University before initiating experiments. Both WT and KO mice demonstrated similar levels of PMNs in bone marrow and whole blood (supplemental Figure 1, available on the Blood Web site), as measured on the Sysmex XN-350 (Kungsbacka, Sweden). All animal studies were approved by the animal ethics committee of Lund University in accordance with the guidelines of the Swedish National Board of Agriculture and the European Union directive for the protection of animals used in science.
Antibodies and reagents
Antibodies for induction of TRALI in mice were 34-1-2s (monoclonal mouse immunoglobulin G2a [IgG2a], which reacts with murine H-2Kd and H-2Dd MHC class I molecules) and AF6-88.5.5.3 (monoclonal mouse IgG2a, which reacts to murine MHC class I H-2Kb), as well as the in vivo CD4+ T-cell–depleting antibody GK1.5 (anti-CD4, rat IgG2b) and the anti-mouse/human CD44 antibody IM7 and its isotype control rat IgG2b LTF-2, all purchased from Bio X Cell (West Lebanon, NH). BM-8 (anti-mouse F4/80–fluorescein isothiocyanate) was purchased from BioLegend (San Diego, CA). RM4-5 (anti-mouse CD4-phycoerythrin) was purchased from BD Pharmingen (San Diego, CA). For OPN-immunohistochemistry staining, rabbit anti-mouse OPN (clone 0-17) was purchased from IBL International (Gunma, Japan) and F(ab′)2 donkey anti-rabbit IgG (H+L)–horseradish peroxidase (HRP)-conjugated was purchased from Jackson ImmunoResearch (West Grove, PA). Anti-OPN antibody, normal goat IgG isotype control antibody, and recombinant mouse OPN were purchased from R&D Systems (Minneapolis, MN). LPS (Escherichia coli O55:B5) and cystamine dihydrochloride were obtained from Sigma-Aldrich Sweden AB (Stockholm, Sweden). Clodronate liposomes were purchased from Liposoma (Amsterdam, The Netherlands).
Antibody-mediated murine TRALI model
TRALI was induced as previously described.4,20 Briefly, 18 hours before TRALI induction, mice were primed with 0.1 mg/kg LPS intraperitoneally and injected intraperitoneally with the monoclonal antibody GK1.5 (4.5 mg/kg) to deplete CD4+ T cells. In indicated experiments, mice were additionally treated with 200 µL of clodronate liposomes intraperitoneally 4 days and 18 hours before TRALI induction. On the day of the experiment, LPS-primed and CD4-depleted (and in indicated experiments also macrophage-depleted) mice were injected IV with 600 µL of a mixture of the TRALI-inducing antibodies 34-1-2s (45 mg/kg) and AF6-88.5.5.3 (4.5 mg/kg) with or without addition of the indicated treatments including recombinant OPN (0.9 mg/kg), isotype antibody (normal goat IgG), anti-OPN antibody (2.25 mg/kg), anti-CD44 antibody IM7 (9 mg/kg), rat IgG2b isotype control LTF-2, and cystamine (50 mg/kg). Subsequently, rectal temperatures were taken every 30 minutes (up to 90 minutes) using a RET-3 rectal probe for mice (VWR International, Mississauga, ON, Canada) connected to a traceable digital thermometer (Physitemp Instruments, Inc, Clifton, NJ). Ninety minutes after TRALI initiation, mice were anesthetized with a mixture of ketaminol and rompun and exsanguinated by cardiac puncture with blood drawn into 100 μL of phosphate-buffered saline (PBS)/citrate phosphate dextrose adenine.
Tissue processing
After the mice were euthanized, the left lung and spleen were harvested and single-cell suspensions were made by manual homogenization of the tissues in PBS/citrate phosphate dextrose adenine followed by transfer of the cells through a 40-μM cell strainer (BD Biosciences, Bedford, MA). Red blood cells in the filtered solution were lysed by addition of 14 mL of an ammonium chloride/potassium bicarbonate lysis solution (0.15 M NH4Cl, 10 mM KHCO3, Na2EDTA; pH 7.2-7.4) for 7 minutes on ice and washed with PBS. The spleen cells were used to verify CD4+ T-cell depletion by flow cytometry.
Lung W/D weight ratios
Lung wet/dry (W/D) weight ratios, a direct measure of pulmonary edema, were determined as previously described.4,13,20 Briefly, the right lung of each mouse was removed and weighed to determine the wet weight and then dried in an oven for 48 hours at 60°C. The dried lung tissues were reweighed to obtain the dry weight. The lung W/D weight ratio was calculated by the formula: net wet weight/net dry weight.
Pulmonary PMN enumeration
The red blood cell–lysed lung cell suspensions were centrifuged onto microscope slides (Superfrost Plus; Fisher Scientific, Pittsburgh, PA) using a Centurion Scientific PrO-Cyt LCD centrifuge (Chichester, United Kingdom) followed by staining with Hemacolor rapid staining of the blood smear staining kit for microscopy (Merck KGaA, Darmstadt, Germany). The slides were mounted with Neo-Mount (Merck KGaA) and imaged using an Olympus BX60F microscope with an SC50 camera in a blinded manner. All nucleated cells were scored in 4 randomly selected and nonoverlapping fields using ImageJ software, and the pulmonary PMN percentage was obtained by the formula: average number of observed PMN (in 4 fields)/average number of nucleated cells (in 4 fields) × 100.
Lung tissue histology and immunohistochemistry
A segment of the left lung was fixed in Histofix (Histolab Products AB, Askim, Sweden) and embedded into paraffin and sectioned (3 µm) with a microtome. The tissue sections were placed on slides (Superfrost Plus; Fisher Scientific) and deparaffinized in serial baths of xylene and ethanol followed by staining using Mayer hematoxylin and 0.2% eosin (Histolab Products AB, Askim, Sweden) or immunohistochemistry staining. Lung histology was quantitatively scored for lung injury as previously described.21 For the immunohistochemistry staining of OPN, endogenous peroxidase was blocked with 3% H2O2 (Boster Biological technology, Pleasanton, CA) followed by heat-induced epitope retrieval in citrate buffer pH6 (Boster Biological Technology, Pleasanton, CA). Subsequently, blocking was performed using 5% bovine serum albumin and 0.05% Triton-X 100 (Sigma-Aldrich Chemie GmbH, Steinheim, Germany) in PBS (Medicago AB, Uppsala, Sweden) and the primary antibody rabbit anti-mouse OPN (clone: 0-17; IBL International) was incubated at 10 µg/mL overnight at 4°C. The slides were washed in PBS and incubated with the secondary antibody F(ab′)2 donkey anti-rabbit IgG (H+L)-HRP-conjugated (Jackson ImmunoResearch) at a 1/2000 dilution for 45 minutes at room temperature. Tissue sections were stained with a DAB Chromogenic Substrate kit (Boster Biological Technology) according to the manufacturer’s instructions. The slides were counterstained with Mayer hematoxylin (Histolab Products AB) and mounted with Neo-Mount (Merck KGaA). The stained slides were imaged using an Olympus BX60F microscope with an SC50 camera.
Cytokine measurements
Blood obtained via cardiac puncture was centrifuged at 2500g for 15 minutes (without brake) and plasma was collected, aliquoted, and stored at −80°C for subsequent batch analysis. The thawed plasma was analyzed for murine macrophage inflammatory protein-2 (MIP-2) or OPN levels using solid-phase sandwich enzyme-linked immunosorbent assay (ELISA) kits: the mouse CXCL2/MIP-2 Quantikine ELISA kit or mouse OPN ELISA Duoset, respectively (R&D Systems). Additionally, other mentioned cytokines, including IL-6, were evaluated using the Bio-Plex Pro Mouse Cytokine 23-plex assay (Bio-Rad, Hercules, CA). All ELISAs were performed according to the manufacturer’s protocol.
Statistical analysis
All hypotheses were made before initiation of an experiment and it was predecided whether 1-tailed or 2-tailed statistical testing would be applied. Normal distribution was evaluated and consequently the appropriate (non)parametric statistical tests were selected. Statistical analysis was carried out using GraphPad Prism 7.04 software for Windows (GraphPad Software, San Diego, CA) with statistical significance set at P < .05. The specific statistical tests used are mentioned in the figure legends. All error bars in the manuscript represent standard deviation (SD).
Results
OPN KO mice are resistant to antibody-mediated PMN-dependent TRALI
We first validated our previously established murine C57BL/6 TRALI model,4,20 by priming the SPF-housed mice with low-dose LPS and depleting their CD4+ T cells, prior to initiating TRALI with infusion of anti-MHC class I antibodies (34-1-2s and AF6-88.5.5.3). Compared with untreated mice, C57BL/6 WT mice suffered from antibody-mediated TRALI as shown by their significantly increased lung W/D weight ratio (4.72 vs 4.50, respectively; P < .0001; Figure 1A). This also corresponded to significantly increased levels of pulmonary PMNs compared with untreated naive mice (34% vs 5%; P < .0001; Figure 1B). In contrast to the WT mice, however, C57BL/6 OPN (KO) mice were resistant to antibody-mediated TRALI induction as they did not display any significant increase in lung W/D levels or pulmonary PMN accumulation compared with naive OPN KO mice (lung W/Ds, 4.83 vs 4.75, and pulmonary PMNs, 18% vs 16%, respectively; Figure 2).

Antibody-mediated TRALI is associated with increased pulmonary PMN accumulation in WT mice. Lung W/D weight ratios (A) and percentage of pulmonary PMNs (B) in C57BL/6 WT mice primed with LPS, depleted of CD4+ T cells, and injected (or not) with anti-MHC class I antibodies (clones 34-1-2s and AF6-88.5.5.3). Statistical analysis was performed with a 1-tailed unpaired Student t test. Each dot represents 1 mouse and error bars represent SD. ****P < .0001.
Antibody-mediated TRALI is associated with increased pulmonary PMN accumulation in WT mice. Lung W/D weight ratios (A) and percentage of pulmonary PMNs (B) in C57BL/6 WT mice primed with LPS, depleted of CD4+ T cells, and injected (or not) with anti-MHC class I antibodies (clones 34-1-2s and AF6-88.5.5.3). Statistical analysis was performed with a 1-tailed unpaired Student t test. Each dot represents 1 mouse and error bars represent SD. ****P < .0001.

OPN KO mice are resistant to antibody-mediated TRALI, however, OPN administration restores PMN-dependent antibody-mediated TRALI. Lung W/D weight ratios (A) and percentage of pulmonary PMNs (B) in C57BL/6 OPN KO mice primed with LPS, depleted of CD4+ T cells, and injected with anti-MHC class I antibodies (clones 34-1-2s and AF6-88.5.5.3) and either PBS or OPN. For statistical analyses, only significant comparisons of interest are shown. Statistical analysis was performed with a 1-way analysis of variance (ANOVA) with a Tukey post hoc test. Each dot represents 1 mouse and error bars represent SD. *P < .05, ****P < .0001. NS, nonsignificant.
OPN KO mice are resistant to antibody-mediated TRALI, however, OPN administration restores PMN-dependent antibody-mediated TRALI. Lung W/D weight ratios (A) and percentage of pulmonary PMNs (B) in C57BL/6 OPN KO mice primed with LPS, depleted of CD4+ T cells, and injected with anti-MHC class I antibodies (clones 34-1-2s and AF6-88.5.5.3) and either PBS or OPN. For statistical analyses, only significant comparisons of interest are shown. Statistical analysis was performed with a 1-way analysis of variance (ANOVA) with a Tukey post hoc test. Each dot represents 1 mouse and error bars represent SD. *P < .05, ****P < .0001. NS, nonsignificant.
OPN administration restores TRALI responses in OPN KO mice
Administration of recombinant OPN to OPN KO mice significantly induced TRALI reactions compared with untreated naive OPN KO mice (lung W/Ds, 5.12 vs 4.75; P < .05; Figure 2A). The TRALI-inducing effect of OPN administration to OPN KO mice was associated with increased levels of pulmonary PMNs (38% vs 16% compared with untreated naive OPN KO; P < .0001; Figure 2B).
OPN blockade with an anti-OPN antibody prevents TRALI in WT mice
Alternatively, in vivo blocking of OPN in WT mice by administration of an anti-OPN antibody demonstrated decreased lung W/Ds as compared with treatment with an isotype antibody (4.48 vs 4.69, respectively; P < .05; Figure 3A). The OPN-blocking response during TRALI was associated with a decreased level of pulmonary PMN accumulation as compared with treatment with an isotype antibody (14% vs 34%, respectively; P < .0001; Figure 3B).

Blockade of OPN in WT mice or macrophage depletion prevents the occurrence of PMN-dependent antibody-mediated TRALI in C57BL/6 WT mice. Lung W/D weight ratios (A,C) and percentage of pulmonary PMNs (B,D) in C57BL/6 WT mice primed with LPS, depleted of CD4+ T cells, injected with anti-MHC class I antibodies (clones 34-1-2s and AF6-88.5.5.3), either isotype-control antibody or anti-OPN antibody, and depleted of macrophages (C-D) or not (A-B). Statistical analysis was performed with a 1-tailed unpaired Student t test. Each dot represents 1 mouse, and error bars represent SD. *P < .05, ****P < .0001.
Blockade of OPN in WT mice or macrophage depletion prevents the occurrence of PMN-dependent antibody-mediated TRALI in C57BL/6 WT mice. Lung W/D weight ratios (A,C) and percentage of pulmonary PMNs (B,D) in C57BL/6 WT mice primed with LPS, depleted of CD4+ T cells, injected with anti-MHC class I antibodies (clones 34-1-2s and AF6-88.5.5.3), either isotype-control antibody or anti-OPN antibody, and depleted of macrophages (C-D) or not (A-B). Statistical analysis was performed with a 1-tailed unpaired Student t test. Each dot represents 1 mouse, and error bars represent SD. *P < .05, ****P < .0001.
Macrophage depletion prevents PMN-dependent TRALI in WT mice
To assess the role of macrophages in TRALI, we depleted macrophages in WT mice using clodronate liposomes (supplemental Figure 2). Subsequently, we induced TRALI by LPS priming and CD4+ T-cell depletion followed by infusion of TRALI antibodies, with co-infusion of either isotype antibody or anti-OPN antibody. When macrophages were depleted, no TRALI occurred as there was no significant difference in lung W/Ds with the co-infusion of isotype antibody vs anti-OPN antibody (Figure 3C). Accordingly, when macrophages were depleted and TRALI was induced, there was also no significant difference in pulmonary PMN numbers upon co-infusion of isotype antibody vs anti-OPN antibody (Figure 3D). This supports a pathogenic role for macrophages in PMN-dependent antibody-mediated TRALI.
OPN induces lung tissue damage in antibody-mediated PMN-dependent TRALI
We next verified the acute lung injury in the various treatment groups by performing lung tissue histology analyses. Compared with naive or PBS-injected control mice (Figure 4A-B, respectively), we observed signs of severe antibody-mediated acute lung injury (increased alveolar septal thickening, exudates indicating edema and intra-alveolar PMN infiltration) in the OPN KO mice injected with recombinant OPN (Figure 4C). On the other hand, no signs of antibody-mediated acute lung injury were observed in WT mice injected with anti-OPN antibody (Figure 4F) or in naive control mice (Figure 4D) in contrast to isotype antibody–injected WT mice, which demonstrated severe acute lung injury (Figure 4E). These data were in full agreement with a quantitative assessment according to the recommended published guidelines for experimental acute lung jury in animals.21 (supplemental Figure 3).
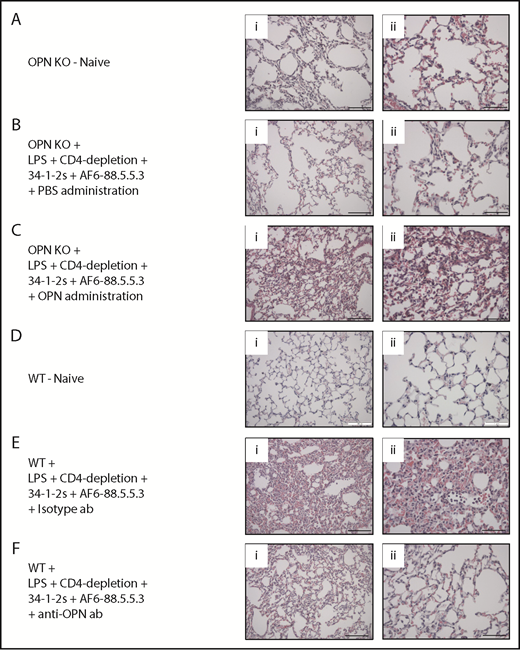
Lung tissue histology of OPN-related TRALI responses. (A-F) Histology performed on lung tissue from the indicated mouse groups. (i-ii) H&E-stained lung tissue images taken at original magnification (i) ×20 and (ii) ×40. Representative images of each indicated group are shown. Scale bars, (i) 100 µm and (ii) 50 µm. ab, antibody.
Lung tissue histology of OPN-related TRALI responses. (A-F) Histology performed on lung tissue from the indicated mouse groups. (i-ii) H&E-stained lung tissue images taken at original magnification (i) ×20 and (ii) ×40. Representative images of each indicated group are shown. Scale bars, (i) 100 µm and (ii) 50 µm. ab, antibody.
Increased pulmonary OPN levels are present in mice suffering from TRALI
As the TRALI-inducing effect of OPN administration corresponded to increased numbers of pulmonary PMNs and OPN is known to regulate recruitment and migration of PMNs,22 we performed lung immunohistochemistry to investigate lung OPN levels during TRALI. In OPN KO mice, which suffered from TRALI due to administration of OPN, we could specifically detect the presence of OPN in the lungs (Figure 5D). This pulmonary staining of OPN did not occur upon LPS priming and CD4+ T-cell depletion (Figure 5A) and was specific to TRALI lungs as also no OPN was detected in the lungs of TRALI-resistant OPN KO mice (Figure 5B) or in lungs using the secondary antibody alone for staining (Figure 5C).
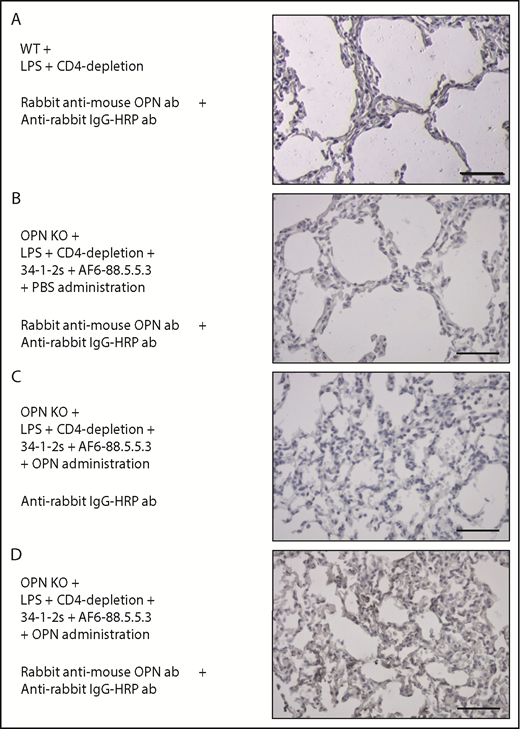
OPN is present in murine TRALI lungs. (A-D) Detection of OPN in the lungs using immunohistochemistry in the indicated mouse groups. Panels on the right represent lung tissue images taken at original magnification ×40 with specific antibody stain after the indicated immunohistochemistry treatments. Scale bars, 50 µm.
OPN is present in murine TRALI lungs. (A-D) Detection of OPN in the lungs using immunohistochemistry in the indicated mouse groups. Panels on the right represent lung tissue images taken at original magnification ×40 with specific antibody stain after the indicated immunohistochemistry treatments. Scale bars, 50 µm.
Plasma OPN levels are low during TRALI but increase upon TRALI prevention with anti-OPN antibody treatment
As we could specifically detect OPN in TRALI lungs, we investigated whether the OPN levels in the plasma would be low during TRALI. We observed that upon TRALI induction, plasma OPN levels were low, however, when TRALI was prevented by anti-OPN antibody treatment, the levels of OPN in the plasma significantly increased as compared with isotype antibody treatment (62 vs 34 ng/mL; P < .0001; Figure 6A). Notably, there was no significant difference in plasma OPN levels of naive mice compared with mice depleted of CD4+ T cells and/or primed with low-dose LPS (Figure 6A).
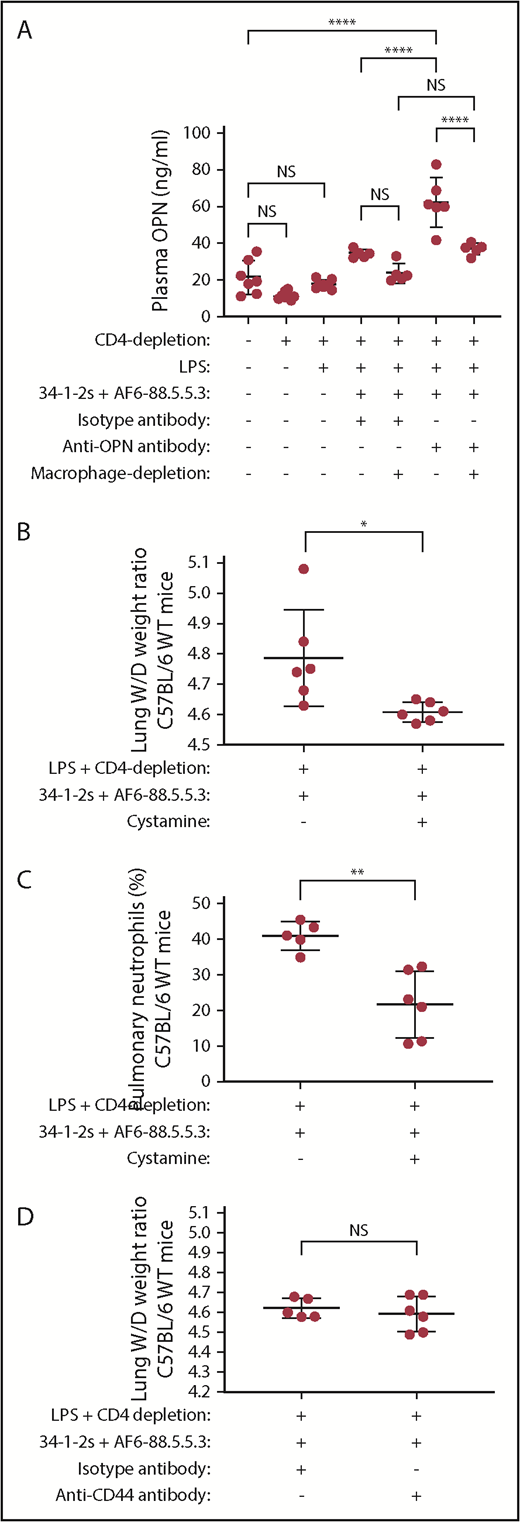
Plasma OPN levels are low during TRALI, increase upon TRALI prevention by OPN blockade with levels remaining low when macrophages are depleted, and treatment with cystamine but not with anti-CD44 antibody prevents antibody-mediated PMN-dependent TRALI. Plasma OPN levels in C57BL/6 WT mice primed with LPS and/or depleted of CD4+ T cells, injected with anti-MHC class I antibodies (clones 34-1-2s and AF6-88.5.5.3), injected with either isotype control antibody or anti-OPN antibody, and depleted or not of macrophages (A). Lung W/D weight ratios (B,D) and percentage of pulmonary PMNs (C) in C57BL/6 WT mice primed with LPS, depleted of CD4+ T cells, injected with anti-MHC class I antibodies (clones 34-1-2s and AF6-88.5.5.3) and either cystamine or not (B,C) or anti-CD44 antibody or isotype antibody (D). Statistical analysis was performed with 1-way ANOVA with a Tukey post hoc test (A) or with 1-tailed unpaired Student t test (B-D). Each dot represents 1 mouse, and error bars represent SD. *P < .05, **P < .01, ****P < .0001.
Plasma OPN levels are low during TRALI, increase upon TRALI prevention by OPN blockade with levels remaining low when macrophages are depleted, and treatment with cystamine but not with anti-CD44 antibody prevents antibody-mediated PMN-dependent TRALI. Plasma OPN levels in C57BL/6 WT mice primed with LPS and/or depleted of CD4+ T cells, injected with anti-MHC class I antibodies (clones 34-1-2s and AF6-88.5.5.3), injected with either isotype control antibody or anti-OPN antibody, and depleted or not of macrophages (A). Lung W/D weight ratios (B,D) and percentage of pulmonary PMNs (C) in C57BL/6 WT mice primed with LPS, depleted of CD4+ T cells, injected with anti-MHC class I antibodies (clones 34-1-2s and AF6-88.5.5.3) and either cystamine or not (B,C) or anti-CD44 antibody or isotype antibody (D). Statistical analysis was performed with 1-way ANOVA with a Tukey post hoc test (A) or with 1-tailed unpaired Student t test (B-D). Each dot represents 1 mouse, and error bars represent SD. *P < .05, **P < .01, ****P < .0001.
Macrophages are the likely source of OPN during TRALI
When macrophages were depleted in vivo and TRALI was prevented by anti-OPN antibody treatment, the levels of plasma OPN were significantly lower as compared with when macrophages were undepleted (37 vs 62 ng/mL; P < .0001; Figure 6A), suggesting that macrophages may be the source of OPN during TRALI.
OPN polymerization appears to be required for PMN-dependent antibody-mediated TRALI, unlike CD44
To investigate the contribution of OPN polymerization in the induction of TRALI, we inhibited the polymerization in vivo using cystamine. Administration of cystamine prevented the onset of TRALI compared with untreated TRALI mice (lung W/Ds, 4.60 vs 4.79; P < .05; Figure 6B). This effect of cystamine treatment was also associated with decreased levels of pulmonary PMNs as compared with untreated TRALI mice (22% vs 41%; P < .01, Figure 6C). Next, we examined whether the leukocyte OPN receptor CD44 was involved in the TRALI response. For that purpose, we blocked CD44 in the murine TRALI model using an anti-CD44 antibody. Blocking CD44, however, did not affect the TRALI response as compared with an isotype antibody (Figure 6D), suggesting that OPN does not engage CD44 in the mediation of antibody-mediated PMN-dependent TRALI.
OPN-induced PMN-dependent antibody-mediated TRALI responses are independent of both MIP-2 and IL-6
As the PMN chemoattractant MIP-2 has previously been described to be upregulated in murine antibody-mediated TRALI,4,13,20,23,24 we determined whether the ability of OPN to induce antibody-mediated PMN-dependent TRALI was related to the levels of MIP-2. We found that compared with naive controls, plasma MIP-2 levels were significantly increased in mice that were infused with the TRALI-inducing antibodies (Figure 7A-B), however, injection or blocking of OPN did not affect the high MIP-2 levels (Figure 7A-B). This indicates that the OPN-related PMN response in TRALI is a distinct mechanism that is independent of plasma MIP-2 levels.
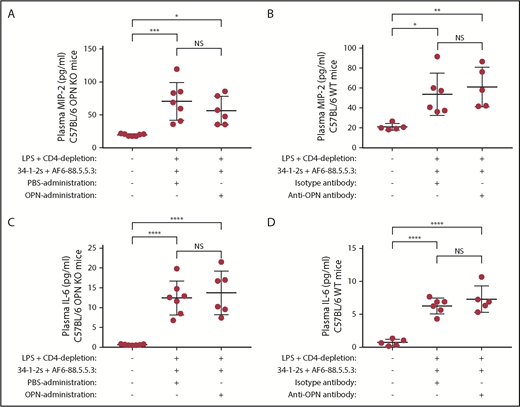
Plasma MIP-2 or IL-6 levels do not change upon OPN-mediated TRALI induction or prevention. Plasma MIP-2 levels (A-B) or IL-6 levels (C-D) in C57BL/6 OPN KO (A,C) or C57BL/6 WT mice (B,D) primed with LPS, depleted of CD4+ T cells and injected with anti-MHC class I antibodies (clones 34-1-2s and AF6-88.5.5.3) and either with PBS or OPN (A,C) or isotype control antibody or anti-OPN antibody (B,D). For statistical analyses, only significant comparisons of interest are shown. Statistical analysis was performed with 1-way ANOVA with a Tukey post hoc test. Each dot represents 1 mouse, and error bars represent SD. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Plasma MIP-2 or IL-6 levels do not change upon OPN-mediated TRALI induction or prevention. Plasma MIP-2 levels (A-B) or IL-6 levels (C-D) in C57BL/6 OPN KO (A,C) or C57BL/6 WT mice (B,D) primed with LPS, depleted of CD4+ T cells and injected with anti-MHC class I antibodies (clones 34-1-2s and AF6-88.5.5.3) and either with PBS or OPN (A,C) or isotype control antibody or anti-OPN antibody (B,D). For statistical analyses, only significant comparisons of interest are shown. Statistical analysis was performed with 1-way ANOVA with a Tukey post hoc test. Each dot represents 1 mouse, and error bars represent SD. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Similarly, we investigated whether the ability of OPN to induce antibody-mediated PMN-dependent TRALI was related to the levels of IL-6, also known to be increased in TRALI patients.9,10 Compared with naive controls, plasma IL-6 levels were significantly increased in mice that were infused with the TRALI-inducing antibodies (Figure 7C-D), however, injection or blocking of OPN did not affect the high IL-6 levels (Figure 7C-D). This also indicates that the OPN-related PMN response in TRALI is a distinct mechanism unrelated to plasma IL-6 levels.
We also assessed whether other PMN chemoattractant cytokines could be involved in antibody-mediated TRALI induction by OPN. We measured plasma levels of granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), interferon-γ, keratinocyte chemoattractant (KC), monocyte chemoattractant protein-1 (MCP-1), MIP-1α, MIP-1β, regulated upon activation normal T-cell expressed and secreted (RANTES), and tumor necrosis factor-α (TNF)-α in OPN KO mice administered OPN (supplemental Figure 4) and in WT mice treated with anti-OPN antibody (supplemental Figure 5). Compared with naive controls, infusion of TRALI antibodies into OPN KO mice significantly increased the plasma levels of GM-CSF, G-CSF, KC, MCP-1, MIP-1α, MIP-1β, RANTES, and TNFα (supplemental Figure 4a-b,d-i). Importantly, however, administration of OPN did not affect any of the tested chemoattractants (supplemental Figure 4), indicating independence of these cytokines from the effects induced by OPN. Similarly, in the WT mice injected with TRALI antibodies, significantly increased plasma levels of G-CSF, MCP-1, KC, MIP-1α, MIP-1β, and RANTES were observed compared with naive control mice (supplemental Figure 5b,d-h). Although treatment with anti-OPN antibody further increased the levels of KC and MIP-1α (supplemental Figure 5e-f), none of the investigated cytokines decreased (supplemental Figure 5). These cytokine data are summarized in supplemental Table 1.
Discussion
Although the pathophysiology of TRALI is incompletely understood, PMNs are assumed to be the major pathogenic cells mediating lung damage based on both human autopsy reports and animal models.1-3 The critical involvement of PMNs in the induction TRALI has been illustrated by murine studies where in vivo depletion of PMNs was shown to result in complete protection from antibody-mediated TRALI.4,25,26
Mechanisms regulating PMN migration toward the lungs in TRALI have not been extensively investigated although multiple murine TRALI models have demonstrated that the PMN chemoattractant MIP-2 plays an important role in this process.4,13,20,23,24 MIP-2 is the murine homolog of human IL-8, which has been shown to be increased in TRALI patients.9,10 On the other hand, OPN is also recognized as a proinflammatory cytokine16 and is one of the most abundantly expressed proteins in various pulmonary disorders including inflammatory, fibrotic, vascular, and malignant lung disorders.17 In addition, OPN is a key cytokine involved in regulating the migration of immune cells,16 particularly the recruitment and migration of PMNs.22
We therefore investigated the role of OPN in regulating pulmonary PMN recruitment in TRALI using a previously established anti-MHC class I antibody-mediated C57BL/6 mouse model of TRALI.4,20 We found that in WT mice, TRALI occurred as expected, however, OPN KO mice were fully protected from TRALI development. Furthermore, injection of purified OPN into the OPN KO mice could restore the pathogenic TRALI response, whereas blocking OPN in WT mice with an anti-OPN antibody prevented the onset of TRALI. All of the OPN-induced TRALI responses were directly related to increased pulmonary PMN recruitment, and the development of acute lung injury was verified by increased lung W/D weight ratios and lung tissue histology analysis. Inflammation is an important risk factor for TRALI development2 and, importantly, OPN has been shown to be upregulated at sites of inflammation such as injured lung epithelium and inflamed vascular endothelium.27,28 We therefore investigated and confirmed the presence of OPN in the lungs of TRALI mice through immunohistochemistry, with no specific detection of OPN upon LPS priming and CD4+ T-cell depletion. The presence of OPN in the lungs coincided with pulmonary PMN accumulation and the occurrence of acute lung injury. Interestingly, the pulmonary presence of OPN during TRALI corresponded to low levels of OPN in the plasma during TRALI as well as to increased levels of plasma OPN upon TRALI prevention with anti-OPN antibody treatment. This suggests that the anti-OPN antibody may retain OPN in the plasma and prevents its migration toward the lungs during TRALI. This suggests that in antibody-mediated TRALI, OPN acts as a proinflammatory cytokine that localizes to the lungs where it recruits PMNs that have been previously shown to mediate lung injury via reactive oxygen species (ROS) production.4
PMNs are known to be poor producers of OPN,22 and OPN is also not expressed in circulating monocytes but is dramatically upregulated during macrophage differentiation with OPN being one of the major factors produced by macrophages.29 We therefore hypothesized that macrophages may be the cellular source of OPN in TRALI. We found that macrophages are indeed pathogenic in murine antibody-mediated PMN-dependent TRALI. Regarding OPN, we observed that macrophage depletion not only prevented pulmonary PMN accumulation and TRALI, but it also prevented increased levels of OPN in plasma, which we observed to occur when the TRALI response was blocked by anti-OPN antibody treatment in the presence of macrophages.
Of interest, OPN has been shown to undergo polymerization in vivo, which was found to be essential for the induction of PMN chemotaxis.30 It was demonstrated that if OPN polymerizes in vivo, the plasma levels of intact OPN decrease compared with polymerization-incompetent OPN.30 Thus, in the TRALI experiments, where anti-OPN-blocking antibody was infused to prevent pulmonary PMN accumulation and TRALI, it may have inhibited OPN polymerization leading to a disruption of the PMN-chemotactic signal and increased levels of intact OPN in the plasma. Of interest, anti-OPN antibody treatment also attenuated PMN migration in a model of sepsis-induced acute lung injury.31 Indeed, we found polymerization of OPN to be required for the induction of antibody-mediated PMN-dependent TRALI, as administration of cystamine, a transglutaminase inhibitor known to block polymerization in vivo,30 prevented pulmonary PMN accumulation and the onset of TRALI.
OPN is able to interact with many cell types via various cell surface receptors, including CD44.32 As the CD44 family of surface receptors regulates adhesion and migration and is one of the major receptors for OPN, we investigated its contribution in mediating TRALI. We blocked CD44 in the TRALI mouse model using an anti-CD44 antibody, but we found that this did not affect the occurrence of TRALI. Alternatively, OPN has been shown to interact with a large variety of integrins, including αvβ1, αvβ3, αvβ5, αvβ6, α8β1, α5β1, α9β1, and α4β7.33-40 Interestingly, polymeric OPN was demonstrated to function as a potent PMN chemoattractant and was shown to interact with integrin α9β1 on PMNs.41 Subsequent research will establish whether, during TRALI, OPN may be interacting with integrins.
Our and other studies have described OPN as being important for the recruitment and migration of PMNs, whereas other neutrophil functions such as phagocytosis and generation of ROS are described as not being affected by OPN.22 In TRALI, it appears that OPN critically recruits PMNs to the lungs and, previously, we showed the critical requirement for PMNs and ROS (using gp91phox KO mice) in antibody-mediated TRALI induction.4 Priming by OPN of neutrophil functions such as ROS production, however, cannot be excluded.
We found that plasma MIP-2 and IL-6 levels were increased in mice that were infused with anti-MHC class I antibodies as compared with naive controls but administration or blocking of OPN did not affect these MIP-2 or IL-6 levels. Thus, OPN appears to be a novel factor regulating pulmonary PMN recruitment and TRALI induction, which is not only critically required but also appears to be functioning independently of MIP-2 and IL-6. We also investigated the effect of OPN administration on other cytokines that have been described to be able to promote PMN chemotaxis (GM-CSF, G-CSF, interferon-γ, KC, MCP-1, MIP-1α, MIP-1β, RANTES, and TNF-α).42-50 None of these cytokines were increased by the administration of OPN, indicating that the pulmonary PMN recruitment and TRALI induction by OPN are distinct mechanisms. This was further supported by demonstrating that these cytokines were not affected in mice where the TRALI response was blocked by infusion of the anti-OPN antibody. None of these cytokines demonstrated decreased levels upon anti-OPN antibody treatment, which prevented the occurrence of pulmonary PMN recruitment and TRALI induction.
Collectively, these data indicate OPN as a novel and critical pathogenic factor that enhances antibody-mediated murine TRALI through stimulation of PMN migration toward the lungs independently of MIP-2, IL-6, or other PMN attractants. This suggests that blocking OPN (using an anti-OPN antibody) may prevent TRALI by impairing pulmonary PMN accumulation, and this should be explored as a potential therapeutic strategy to combat this serious adverse complication of blood transfusion.
Presented as an oral abstract at the 60th annual meeting and exposition of the American Society of Hematology, San Diego, CA, 1-4 December 2018.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by grants from Lund University, Crafoordska Stiftelsen (#20170829), Vetenskapsrådet (Swedish Research Council, VR, #2017-01779), and Avtal om Läkarutbildning och Forskning (ALF).
Authorship
Contribution: R.K. designed and performed experiments, collected, analyzed, and interpreted data, and wrote and edited the paper; G.K. and J.R. performed experiments and collected data; A.E. supplied OPN KO mice and edited the manuscript; and J.W.S. provided financial resources and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for R.K. is Department of Experimental Immunohematology, Sanquin Research, and Landsteiner Laboratory, Amsterdam UMC, University of Amsterdam, Amsterdam, The Netherlands.
The current affiliation for G.K. is Bioscience COPD/IPF, Early Respiratory, Inflammation and Autoimmunity, R&D Biopharmaceuticals, Gothenburg, Sweden.
Correspondence: John W. Semple, Lund University, BMC C14, Klinikgatan 26, 221 84, Lund, Sweden; e-mail: john_w.semple@med.lu.se.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal