

In this issue of Blood, identified abolishment of colony-stimulating factor 1 (CSF1)–mediated supportive paracrine signaling as a potential novel therapeutic strategy in acute myeloid leukemia (AML). “Famine makes greater havoc in an army than the enemy, and is more terrible than the sword.” This maxim, penned by the Roman writer Vegetius, describes the Roman military principle of depleting enemy resources to increase one’s likelihood of victory. Although this approach was developed for wars involving troops and sieges, Edwards et al have identified a mechanism by which it could be usefully applied to treating AML.1
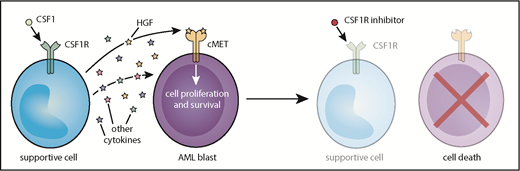
Model of CSF1R inhibitor sensitivity in primary AML patient samples resulting from paracrine secretion of cytokines by CSF1R-expressing supportive cells.
Model of CSF1R inhibitor sensitivity in primary AML patient samples resulting from paracrine secretion of cytokines by CSF1R-expressing supportive cells.
The bone marrow microenvironment has long been suspected to play a role in AML pathogenesis and protection from therapy.2 Clinically, patients with clearance of circulating leukemic blasts after treatment are frequently found to harbor significant residual bone marrow disease, suggesting that bone marrow environment factors play a key role in shielding AML blasts from the cytotoxic effects of therapy. However, efforts to improve clinical responses to chemotherapy by mobilizing blasts out of the bone marrow environment have proved to be largely unsuccessful.3 In the case of targeted therapy, endogenous cytokines such as FLT3 ligand4 and FGF5 have been implicated in resistance to small-molecule FLT3 inhibitors, but the source of these protective cytokines has not been definitively identified. Regardless, multiple cell types in the bone marrow niche have been implicated in providing a permissive environment for leukemia cell survival and proliferation.6,7 Until now, a role for therapeutic targeting of leukemia-supporting rather than actual leukemia cells has not been previously explored in AML.
In functional genetic and small-molecule screens to identify novel AML targets, Edwards et al uncovered the CSF1 receptor (CSF1R) as a target that reduced cell viability in a large proportion of primary AML samples. Sensitivity to CSF1R inhibition was diminished in relapsed and high-risk patients and correlated with increased overall survival. Surprisingly, CSF1R expression was negligible on AML blasts, and mass cytometry analysis revealed high CSF1R (CSF1Rhi) expression in only a very small subset of cells within each AML patient sample, indicating that CSF1R inhibition does not target the bulk AML blast population. Both hepatocyte growth factor (HGF) and conditioned media from the human stromal line HS-5 rescue primary AML samples from CSF1R inhibition, suggesting that CSF1R signaling from a small population of CSF1Rhi cells facilitates the release of HGF and other unidentified cytokines to support viability of AML blasts.
The origin of these supportive CSF1Rhi cells is less clear. CSF1Rhi cells from both AML and healthy donors often co-express myeloid markers, but comparison of samples from patients with AML and healthy donors reveals that there are phenotypic differences between these populations. In 2 AML patients, sorted CSF1R+ cells exhibited the same genetic mutations as those identified in the bulk tumor sample, a finding which suggests that these cells could represent a tumor subpopulation that needs to be further defined. Regardless, the findings of Edwards et al suggest that targeting of paracrine signaling via CSF1R inhibition could be broadly effective in many AML subtypes (see figure). Importantly, clinical grade CSF1R inhibitors8 have been developed and are tolerable in patients, which paves the road to clinical trials that test the efficacy of CSF1R inhibition in AML.
The optimal role of CSF1R inhibition in AML remains to be defined. The decreased sensitivity of samples from relapsed AML patient to CSF1R targeting suggests that de novo AML may be more dependent on the support of CSF1Rhi cells and that these inhibitors are most appropriately used early in treatment, perhaps in combination with chemotherapy (although, as the authors note, it is likely that such inhibitors would be first tested in a relapsed/refractory AML population). Historically, AML treatment has relied on the targeting of cell intrinsic survival mechanisms. However, to improve treatment of AML, we would be wise to learn from the Romans and add the targeting of leukemia cell extrinsic supportive mechanisms to our therapeutic armamentarium.
Conflict-of-interest disclosure: C.C.S. has received research funding from Plexxikon Inc., Astellas Pharma, FujiFilm Pharmaceuticals, and Revolution Medicines.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal