

Key Points
The high-resolution structure and associated dynamics of the platelet integrin-binding VWF C4 domain is presented.
A possible mechanism of gain-of-function variant affecting the arrangement with neighboring domains is proposed.

Abstract
Von Willebrand factor (VWF) is a key player in the regulation of hemostasis by promoting recruitment of platelets to sites of vascular injury. An array of 6 C domains forms the dimeric C-terminal VWF stem. Upon shear force activation, the stem adopts an open conformation allowing the adhesion of VWF to platelets and the vessel wall. To understand the underlying molecular mechanism and associated functional perturbations in disease-related variants, knowledge of high-resolution structures and dynamics of C domains is of paramount interest. Here, we present the solution structure of the VWF C4 domain, which binds to the platelet integrin and is therefore crucial for the VWF function. In the structure, we observed 5 intra- and inter-subdomain disulfide bridges, of which 1 is unique in the C4 domain. The structure further revealed an unusually hinged 2-subdomain arrangement. The hinge is confined to a very short segment around V2547 connecting the 2 subdomains. Together with 2 nearby inter-subdomain disulfide bridges, this hinge induces slow conformational changes and positional alternations of both subdomains with respect to each other. Furthermore, the structure demonstrates that a clinical gain-of-function VWF variant (Y2561) is more likely to have an effect on the arrangement of the C4 domain with neighboring domains rather than impairing platelet integrin binding.
Introduction
Von Willebrand factor (VWF) is essential to the regulation of hemostasis and thrombosis. At sites of vascular injury, VWF binds to platelets and subsequently promotes their aggregation, thereby mediating adhesion to the damaged vessel wall.1 In addition, VWF serves as a carrier and stabilizer for coagulation factor VIII in the blood circulation.1,2
VWF is a large mosaic protein, composed of 3 type A, 6 type C (VWC), and 4 type D domains, as well as a C-terminal cystine knot domain3 (Figure 1A). These domains are connected with one another like beads on a string, forming a long, linear, rodlike molecule of ∼60 nm in length.4 The monomeric units are organized into multimers ranging from dimers to gigantic concatamers of >20 000 kDa,5,6 via disulfide bonding head to head at the N-termini7 and tail-to-tail at the C-termini.8 Furthermore, its great length and multidomain composition facilitate VWF to sense and respond appropriately to hydrodynamic forces of the blood flow, as well as the physiochemical environment.6,9-11 Dimeric VWF, the smallest repeating unit, can adopt both open and flexible, and zipped-up and closed conformations, depending on factors including pH, temperature, presence of divalent ions, and shear force.10-12 Thus, the conformational states of the minimal dimeric units also regulate the overall length of the concatamers, thereby tuning the hemostatic function of VWF.11
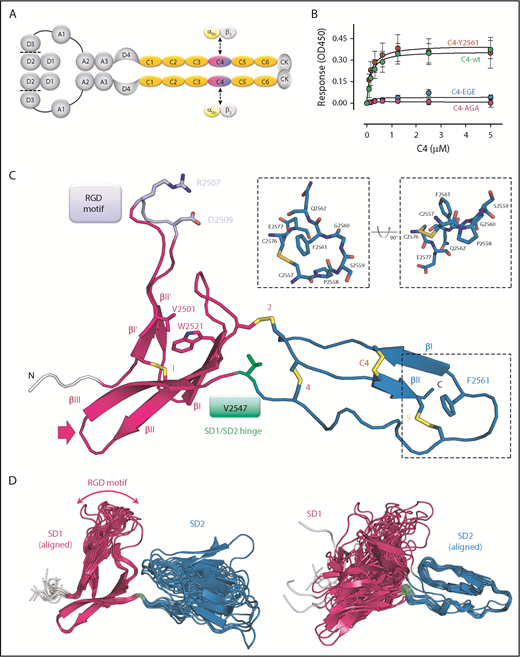
Solution structure of the VWF C4 domain. (A) Schematic presentation of the VWF domain structure with the C4 domain highlighted. The C4 domain has a specific role in binding to the heterodimeric platelet integrin subunits αIIb/β3, as indicated. Prodomain dissociation is indicated by dashed lines. (B) Ability of the VWF C4 domain to bind to αIIbβ3 platelet integrin: wt, Y2561 variant, glutamates (EGE) (R2507ED2509E), and alanines (AGA) (R2507AD2509A). (C) Ribbon diagram of the lowest-energy solution structure of the C4 domain of VWF, with its subdomain structure SD1 (residues 2497-2546) shown in magenta, SD2 (residues 2548-2577) in blue, and the N-terminal cloning tag in light gray. All β-strands are labeled with Roman numbers. Disulfide bridges (compare Table 2) are numbered and colored in yellow. The side chains of other VWF C4 residues described in the text are shown in stick representation with atom-specific colors: V2501 and W2521, magenta; RGD motif (residues 2507-2509), lavender; the SD1/SD2 hinge residue V2547, green; F2561, changed to tyrosine in a clinical variant,15 blue. The extended loop connecting SD1 strands βII and βIII is indicated with an arrow (compare Figure 4). Insets show stick representation of the local environment of F2561. A view rotated by 90° demonstrates that the side chain of F2561 is presented on the surface (right). (D) The 20 lowest-energy NMR conformers are superimposed on SD1 (left) and SD2 (right), respectively, demonstrating substantial SD1/SD2 hinge variability. The flexible RDG motif is indicated.
Solution structure of the VWF C4 domain. (A) Schematic presentation of the VWF domain structure with the C4 domain highlighted. The C4 domain has a specific role in binding to the heterodimeric platelet integrin subunits αIIb/β3, as indicated. Prodomain dissociation is indicated by dashed lines. (B) Ability of the VWF C4 domain to bind to αIIbβ3 platelet integrin: wt, Y2561 variant, glutamates (EGE) (R2507ED2509E), and alanines (AGA) (R2507AD2509A). (C) Ribbon diagram of the lowest-energy solution structure of the C4 domain of VWF, with its subdomain structure SD1 (residues 2497-2546) shown in magenta, SD2 (residues 2548-2577) in blue, and the N-terminal cloning tag in light gray. All β-strands are labeled with Roman numbers. Disulfide bridges (compare Table 2) are numbered and colored in yellow. The side chains of other VWF C4 residues described in the text are shown in stick representation with atom-specific colors: V2501 and W2521, magenta; RGD motif (residues 2507-2509), lavender; the SD1/SD2 hinge residue V2547, green; F2561, changed to tyrosine in a clinical variant,15 blue. The extended loop connecting SD1 strands βII and βIII is indicated with an arrow (compare Figure 4). Insets show stick representation of the local environment of F2561. A view rotated by 90° demonstrates that the side chain of F2561 is presented on the surface (right). (D) The 20 lowest-energy NMR conformers are superimposed on SD1 (left) and SD2 (right), respectively, demonstrating substantial SD1/SD2 hinge variability. The flexible RDG motif is indicated.
Defects or deficiency of VWF can lead to von Willebrand disease, which is the most common inherited bleeding disorder.1,13 Type IIB von Willebrand disease, although classed as gain-of-function (GOF) due to increased affinity for platelet glycoprotein Ibα (GPIbα), still lead to prolonged bleeding.14 New GOF variants have recently been identified to be associated with thromboembolism.15 One such GOF variant is the substitution of F2561 to a tyrosine located in the C4 domain (Y2561).
Despite the well-characterized functions, our current understanding of the molecular structure and thus mechanisms of function of VWF remains poor. The general architecture has been shown at low resolution by electron microscopy.10 Atomic-resolution structures, however, are only available for the 3 A domains,16-18 the C-terminal cystine knot domain domain,19 and the small D′ segment of the D′D3 domain (TIL′E′).20 The entire “stem” region consisting of 6 C domains has not yet been characterized structurally.
The VWF C domain presents one of the most common motifs in extracellular proteins, with 201 VWC domains found in 70 human proteins (http://smart.embl-heidelberg.de/).21 The VWC domain is a small, ∼10-kDa domain, typically of 75 to 100 residues in length. Available structures include the VWC domain of Collagen IIA (Col2a),22,23 CCN3 growth factor,23 and Crossveinless (CV-2),24,25 an inhibitor for Bone Morphogenetic Protein signaling. A subdivision of an N- and C-terminal half is common among all 3 VWC structures, generally associated with an extended, flexible orientation. The structures of these domains have revealed a conserved pattern of 5 disulfide bonds with identical connectivity. Based on these findings, and in the absence of any experimental structural data on the C-terminal stem of the VWF protein, predictions of disulfide linkage in the VWF C domains have previously been made by sequence homology.3
In this study, the first high-resolution solution structure of a VWF C domain is presented. For structure determination, we have selected the C4 domain, which has a unique ability to bind to platelet integrin αIIbβ3 (also known as GPIIb/IIIa).1,26 The C4 structure exhibits a distinct disulfide pattern, different from other known C domains, and reveals a subdomain arrangement that is unusually kinked. In the structure, the platelet integrin-binding loop is, nevertheless, highly exposed, and qualitative binding data to platelet integrin αIIbβ3 confirm its role in the interaction. Our structural findings on the C4 domain suggest that similarly kinked subdomain arrangements from neighboring VWF C domains may have an amplifying effect on the overall VWF C-stem architecture and may explain how this affects binding to platelet integrin.
Materials and methods
Expression, refolding, and purification of VWF C4
Human VWF C4 (residues 2497-2577, UniProt P04275, sequence denoted as “wild-type” [wt], F2561), plus an N-terminal extension of GSMA from the thrombin cleavage site, was recombinantly expressed in BL21(DE3) Escherichia coli using the pHAT3 vector,27 which contains an N-terminal His6-tag. The transformed cells were cultured in isotopically labeled M9 minimal medium at 37°C, induced with 400 μM isopropyl-β-d-thiogalactoside at optical density at 600 nm wavelength of 0.6 to 0.8 for 4 hours. VWF C4 was found in inclusion bodies, which were separated from the soluble fractions by centrifugation upon lysis of the cells. The protein was denatured in 6 M urea and guanidine hydrochloride before refolding by rapid 1:10 dilution into 50 mM sodium phosphate pH 7.5, 0.33 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate, 2 mM reduced glutathione, and 0.1 mM oxidized glutathione. Refolded protein was purified first by nickel-nitrilotriacetic acid affinity chromatography, followed by cleavage of the His-tag by thrombin protease, and by reversed phase chromatography (ACE 5 C18-300), before lyophilization and resuspension in 20 mM sodium phosphate pH 6.5. The C4 Y2561 variant gene was generated by a 2-step polymerase chain reaction protocol using overlapping oligonucleotides across the mutated regions, and the respective protein was produced as for the wt C4 domain, at a similar level of expression and thermal stability (supplemental Figure 2A, available on the Blood Web site).
NMR spectroscopy experiments
All nuclear magnetic resonance (NMR) experiments were carried out at 298 K (except where stated otherwise) on a Bruker Avance III NMR spectrometer equipped with a cryogenic triple resonance gradient probe head at a magnetic field strength corresponding to a proton Larmor frequency of 600 MHz. All spectra were processed with NMRPipe28 and analyzed using Sparky,29 CARA (http://cara.nmr.ch/doku.php), and CCPNMR.30 For backbone assignment, 15N-HSQC, HNCACB, CBCA(CO)NH, HNCA, and HNCO spectra were recorded.31 Side-chain assignments were done using 13C-HSQC, HBHACONH, HCCH-TOCSY, HCCCONH, and CCCONH spectra.31 Distance restraints were obtained from 15N- and 13C-edited 3-dimensional NOESY-HSQC, and 13C-edited HMQC-NOESY experiments.31 Assignment of amide proton/nitrogen backbone resonances of the Y2561 variant were transferred from the wt and confirmed by a 15N-NOESY-HSQC experiment using sequential HN-Hα-1 resonances.
Structure determination and analysis
The solution NMR structure of the VWF C4 domain was determined by automated nuclear Overhauser enhancement (NOE) crosspeak assignment and torsion angle dynamics, using CYANA 3.0.32 Distance restraints from the CYANA calculation and TALOS+33 derived dihedral angle restraints were used in a water refinement calculation using ARIA1.2.34 Structure validation of the final ensemble of 20 structures with lowest energies was done using PROCHECK36 and WHATCHECK.35 The structural statistics are shown in Table 1. Structure-based sequence alignment was performed using PDBeFold (http://www.ebi.ac.uk/msd-srv/ssm),37 and geometric analysis using DynDom (http://fizz.cmp.uea.ac.uk/dyndom/).38
Structural statistics of the VWF C4 domain NMR ensemble
| Experimental restraints | |
| Total NOEs | 2478 (assigned 2255) |
| Distance restraints | 1158 |
| Short range (|i − j| ≤ 1) | 650 |
| Medium range (|i − j| < 5) | 85 |
| Long range (|i − j| > 5) | 423 |
| Dihedral restraints (φ/ψ) | 144 |
| Structural quality | |
| Coordinate precision (Å, residues 2496-2503, 2513-2548) | |
| Backbone (N, Cα, C′) | 0.46 ± 0.16 |
| Heavy atoms | 0.77 ± 0.13 |
| Coordinate precision (Å, residues 2562-2577) | |
| Backbone (N, Cα, C′) | 0.39 ± 0.15 |
| Heavy atoms | 1.10 ± 0.20 |
| Restraint RMSD | |
| Distance restraints, Å | 0.018 ± 0.001 |
| Dihedral restraints, ° | 1.45 ± 0.24 |
| Deviation from idealized geometry | |
| Bond lengths, Å | 0.004 ± 0.0002 |
| Bond angles, ° | 0.52 ± 0.02 |
| Ramachandran analysis, % | |
| Favored regions | 84.7 |
| Allowed regions | 14.0 |
| Generously allowed | 0.0 |
| Disallowed | 1.3 |
| Whatcheck analysis | |
| First-generation packing | −3.061 |
| Second-generation packing | −3.531 |
| Ramachandran plot appearance | −3.266 |
| χ−1/χ−2 rotamer normality | −2.452 |
| Backbone conformation | −0.861 |
| Experimental restraints | |
| Total NOEs | 2478 (assigned 2255) |
| Distance restraints | 1158 |
| Short range (|i − j| ≤ 1) | 650 |
| Medium range (|i − j| < 5) | 85 |
| Long range (|i − j| > 5) | 423 |
| Dihedral restraints (φ/ψ) | 144 |
| Structural quality | |
| Coordinate precision (Å, residues 2496-2503, 2513-2548) | |
| Backbone (N, Cα, C′) | 0.46 ± 0.16 |
| Heavy atoms | 0.77 ± 0.13 |
| Coordinate precision (Å, residues 2562-2577) | |
| Backbone (N, Cα, C′) | 0.39 ± 0.15 |
| Heavy atoms | 1.10 ± 0.20 |
| Restraint RMSD | |
| Distance restraints, Å | 0.018 ± 0.001 |
| Dihedral restraints, ° | 1.45 ± 0.24 |
| Deviation from idealized geometry | |
| Bond lengths, Å | 0.004 ± 0.0002 |
| Bond angles, ° | 0.52 ± 0.02 |
| Ramachandran analysis, % | |
| Favored regions | 84.7 |
| Allowed regions | 14.0 |
| Generously allowed | 0.0 |
| Disallowed | 1.3 |
| Whatcheck analysis | |
| First-generation packing | −3.061 |
| Second-generation packing | −3.531 |
| Ramachandran plot appearance | −3.266 |
| χ−1/χ−2 rotamer normality | −2.452 |
| Backbone conformation | −0.861 |
NMR relaxation
Measurements of R1, R2, and 1H-15N heteronuclear NOE experiments for VWF C4 wt and the Y2561 variant were acquired at static magnetic fields corresponding to proton Larmor frequencies of 500, 600, 700, and 800 MHz at 298 K using standard pulse sequences.39,40 Experiments at 283 K and 310 K were recorded only at 600 MHz. For all R1 experiments, relaxation delays of 20, 50, 100, 150, 250, 400, 500, 650, 800, 1000, 1300, and 1600 milliseconds were used. Duplicate data points for estimation of uncertainties in peak volumes were recorded for the 20-millisecond relaxation delay. All R2 experiments were recorded with relaxation delays of 16, 32, 48, 64, 80, 96, 112, 128, 160, 192, 224, and 256 milliseconds. Duplicate data points were recorded for the 16-milliseconds relaxation delay.
To assess the time scale of dynamics for certain residues Carr-Purcell-Meiboom-Gill (CPMG),41,42 relaxation dispersion experiments at 298 K were recorded at 600 and 800 MHz using a previously published pulse sequence.43 Effective fields, defined as νCPMG = 1/(4τCP), where 2τCP is the interval between refocusing pulses of 33, 66, 100, 133, 200, 266, 333, 400, 466, 533, 600, 666, 800, and 933 Hz, were used. Peak intensities were transformed to effective transverse relaxation rates according to R2,eff = ln(I0)/I(νCPMG)/T, where I(νCPMG) is the intensity for different values of νCPMG, I0 is the intensity without the CPMG sequence block, and T is the duration of the constant time relaxation delay (30 milliseconds). PINT44 was used for peak integration and fits of spin relaxation and CPMG data.
ROTDIF45 was employed to determine the diffusion tensor from the R2/R1 ratios. The data were fitted better to the axially symmetric model than the isotropic model. Using an anisotropic model did not improve the fit. For calculation of the rotational correlation time, residues with heteronuclear NOE values <0.65 were excluded from the analysis.
MD simulations
Simulations employed a previously published protocol46 using GROMACS 5.1.x47 and the CHARMM22* all-atom force field.48 The Verlet scheme was used with standard twin-range 10- to 12-Å cutoffs, utilizing Particle-Mesh Ewald electrostatics and Force-switching van der Waals interactions. Time steps were set at 4 fs using virtual site construction. For the wt C4 domain, each of the 20 NMR conformations was subjected to the following equilibration and production protocols: solubilization in an 8-nm dodecahedron box with periodic boundaries containing 150 mM NaCl, 2500-step energy-minimization by steepest descent, equilibration over 2 nanoseconds using gradually relaxing position restraints, first over the side chains and then over the backbone, and finally 100-nanosecond production simulations. All simulations were maintained under constant number, pressure, and temperature conditions, using velocity-rescaling thermostats (τt = 2.5 ps) and Berendsen barostats (τp = 5.0 ps) during equilibration and then switching to Nosé-Hoover thermostats and Parinello-Rahman barostats during production. The Y2561 variant was derived from the 20 wt structures by manual mutation in visual molecular dynamics (MD).49 Each mutant was equilibrated identically to the wt, followed by 200-nanosecond production simulations, with the first 100 nanoseconds discarded. The computation of NMR spin-relaxation from the 20 × 100 nanoseconds ensembles follows procedures described in Chen et al,50 except where the global tumbling of ROTDIF was used to replace simulated values before fitting to experiment.
Solid-phase binding assay
Human αIIbβ3 was immobilized, and C4 domain variants were in solution. Details of experiments are described in supplemental Methods.
Results
Solution structure of VWF C4 domain
The C4 domain structure, covering residues S2497 to E2577, of the most frequently occurring human VWF sequence variant (F2561), was determined by solution NMR spectroscopy (Figure 1C-D;,Table 1; supplemental Figure 1). The backbone resonances of all residues were assigned except for S2568, A2569, and C2570, which indicate local motions in the intermediate exchange regime. This leads to a total assignment of 96.8% of all nuclei including side chains.
The overall C4 fold is divided into 2 subdomains, SD1 (S2497-N2546) and SD2 (S2548-E2577), marking V2547 as hinge residue (Figure 1C). SD1 is composed of 2 antiparallel β-sheets. The first one comprises 2 short β-strands I′-II′ connected by a long 10-residue hairpin loop, with the αIIbβ3 integrin binding arginine-glycine-aspartatic acid (RGD) motif (residues 2507-2509) at its tip. The absence of medium- and long-range NOEs at this motif and thus larger conformational heterogeneity between the 20 lowest energy structures of the NMR ensemble suggests high mobility (Figure 1D left). This has been further confirmed by 15N relaxation experiments, showing that the RGD loop region exhibits very fast dynamics in the nanosecond-picosecond time scale indicated by decreasing R2/R1 and heteronuclear NOE values (Figure 2). The second SD1 β-sheet comprises 3 antiparallel strands I-II-III. Together, the 2 SD1 β-sheets form a 2-layered structure. Interestingly, the interface of the 2 β-sheets is devoid of any specific hydrogen-bond interactions. The most prominent hydrophobic interaction is established between V2501 from β-strand I′ of the first sheet and W2521 from β-strand I of the second sheet. The C-terminal SD2 comprises one 2-stranded antiparallel β-sheet I-II. The sheet is preceded by a long sequence stretch without any regular secondary structure, connecting the C-terminus of SD1 with the first strand of the SD2 β-sheet.

Structural dynamics within the VWF C4 domain. (A) Experimental R2/R1 values derived from 15N relaxation NMR experiments at 600 MHz. Wt (black) and Y2561 variant (red) show similar profiles, indicating very similar dynamics. At high concentrations (hc, yellow), R2/R1 values increase globally. (B) MD simulation of spin relaxation parameters predicts a very similar profile except for the exchange contribution of V2547. Error bars indicate standard deviation produced from 4 trajectory subchunks. (C) Rotational correlation time (τc) of wt and Y2561 variant at 4 different field strengths. The Y2561 variant shows elevated τc values, which further increase upon elevated concentration. (D) R2 values at 4 different fields for both wt and Y2561 variant are similar and show field dependence of Rex for residue V2547. B0, magnetic field strength.
Structural dynamics within the VWF C4 domain. (A) Experimental R2/R1 values derived from 15N relaxation NMR experiments at 600 MHz. Wt (black) and Y2561 variant (red) show similar profiles, indicating very similar dynamics. At high concentrations (hc, yellow), R2/R1 values increase globally. (B) MD simulation of spin relaxation parameters predicts a very similar profile except for the exchange contribution of V2547. Error bars indicate standard deviation produced from 4 trajectory subchunks. (C) Rotational correlation time (τc) of wt and Y2561 variant at 4 different field strengths. The Y2561 variant shows elevated τc values, which further increase upon elevated concentration. (D) R2 values at 4 different fields for both wt and Y2561 variant are similar and show field dependence of Rex for residue V2547. B0, magnetic field strength.
The C4 structure comprises 5 disulfide bridges (Figure 1C; Table 2). Chemical shift values of Cβ resonances clearly indicated that all cysteines are involved in disulfide bonds51 (supplemental Table 1). Three disulfides are formed by cysteines within 1 of the 2 subdomains SD1 (bridge 1) and SD2 (bridges 5 and C4) and thus are thought to be fold determinants of these subdomains. The latter disulfide bridge (C2565-C2574) is specific to the VWF C4 domain, hence named “C4” bridge (further details below). The 2 remaining disulfide bridges connect the 2 subdomains (bridges 2 and 4) and thus appear to be determinants of the observed SD1/SD2 arrangement.
Disulfide bridge connections in VWF C4
| S-S bridge | Residue (1) | Secondary structure | Residue (2) | Secondary structure | SD1/SD2 hinge determining | Comment |
|---|---|---|---|---|---|---|
| 1 | 2499 | βI′ (SD1) | 2533 | βII (SD1) | No | Intra SD1, canonical |
| 2 | 2528 | Loop βI-βII (SD1) | 2570 | Loop βI-βII (SD2) | Yes | SD1/SD2, canonical |
| 3 | Missing | |||||
| 4 | 2549 | Hinge SD1/SD2 | 2571 | Loop βI-βII (SD2) | Yes | SD1/SD2, canonical |
| 5 | 2557 | 2576 | βII (SD2) | No | SD2, noncanonical | |
| C4 | 2565 | βI (SD2) | 2574 | βII (SD2) | No | C4 only, noncanonical |
| S-S bridge | Residue (1) | Secondary structure | Residue (2) | Secondary structure | SD1/SD2 hinge determining | Comment |
|---|---|---|---|---|---|---|
| 1 | 2499 | βI′ (SD1) | 2533 | βII (SD1) | No | Intra SD1, canonical |
| 2 | 2528 | Loop βI-βII (SD1) | 2570 | Loop βI-βII (SD2) | Yes | SD1/SD2, canonical |
| 3 | Missing | |||||
| 4 | 2549 | Hinge SD1/SD2 | 2571 | Loop βI-βII (SD2) | Yes | SD1/SD2, canonical |
| 5 | 2557 | 2576 | βII (SD2) | No | SD2, noncanonical | |
| C4 | 2565 | βI (SD2) | 2574 | βII (SD2) | No | C4 only, noncanonical |
Comparison of the 20 lowest energy conformers unambiguously identifies a short residue segment centered on V2547 as a flexible hinge, allowing limited flexibility in the orientation between SD1 and SD2. In order to confirm these observations experimentally, 15N relaxation experiments were performed to obtain a residue-wise quantitative picture (Figure 2; supplemental Figure 3). The most prominent peak in the relaxation profile is found for V2547, which confirms that this residue undergoes slow conformational exchange in the microsecond to millisecond time scale. The exchange contribution Rex to R2 is considerable and ranges from 7.7 s−1 to 19.7 s−1 at 500 to 800 MHz, respectively. To assess the time scale of these exchanges, CPMG experiments at 600 and 800 MHz were performed (supplemental Figure 4). As the dispersion profile for residue V2547 and neighboring residues is flat, but retains high R2 values throughout the CPMG frequencies used, we conclude that the conformational change is in the low microsecond range.
The consistently lower R2/R1 values for SD2 indicate higher global mobility compared with SD1 (Figure 2A-B). Due to the flexible hinge, SD2 presumably moves with respect to SD1 and samples different orientations. The global rotational correlation time τc of the C4 domain calculated from R2/R1 values is 7.5 nanoseconds, which agrees well with the value predicted from the NMR ensemble by ROTDIF (7.9 nanoseconds; Figure 2C). Interestingly, the dynamics of the hinge region and especially residue V2547 vary considerably upon temperature changes (supplemental Figure 5). At an elevated, physiological temperature (37°C), the flexibility is increased, and there is no exchange contribution to R2. Dynamics are even slower at 10°C compared with 25°C, indicated by the broadened peak of V2547, which vanishes into the noise. In summary, these NMR analyses demonstrate microsecond-level flexibility between SD1 and SD2 domains that is highly sensitive to temperature variations.
Modeling of C4 domain dynamics
To assist in interpreting the NMR dynamics, multiple MD simulations were conducted to examine possible dependence of domain dynamics upon mutation (Figure 3). Two angular parameters were formulated to define the relative SD1-SD2 arrangements observed. The dihedral twist angle φ is defined by the centers of mass of SD1 and SD2, plus the centers of mass of V2547 and C2528. The complementary tilt angle θ is defined by θ = 180° + θ1 − θ2, where θ1 is defined by the angle (center of gravity SD1)-C2547-C2528 and θ2 C2547-C2528-(center of gravity SD2) (Figure 3A inset). When both angles φ and θ are 180°, the SD1/SD2 arrangement is fully extended. The more both angles deviate from 180°, the more the arrangement becomes twisted and tilted. As a result of the MD simulation, we found a skewed tilt distribution centered at θ ∼190° with a θ range of 140° to 230° (Figure 3A left). The twist angle distribution is more bimodal, where the more populated one peaks at φ ∼170° (cluster I) and a less populated one (cluster II) in a φ angle range of 250° to 290°. In terms of an overall SD1/SD2 arrangement, the second cluster II is significantly more bent than the first cluster I (Figure 3B).

SD1/SD2 arrangement diversity of the VWF C4 domain. (A) Twist/tilt distribution of VWF C4 domain SD1/SD2 arrangements (wt, left; Y2561 variant, right) estimated by MD simulations. The counts of MD models are shown as a heat map and as histograms along the tilt and twist axes. The heat map colors are defined by counts, as indicated on the right. Highly populated SD1/SD2 arrangements (clusters) are indicated with labels I (wt, Y2561), II (wt, Y2561), and III (Y2561 only). A scheme illustrating the definition of twist and tilt angles is inserted, using colors consistent in panel B. (B) Representative examples of SD1/SD2 arrangement clusters I, II, and III are shown, and their approximate population densities are indicated by “+” signs. Centers of mass used for calculating tilt and twist angles are shown as colored spheres and connected by gray sticks.
SD1/SD2 arrangement diversity of the VWF C4 domain. (A) Twist/tilt distribution of VWF C4 domain SD1/SD2 arrangements (wt, left; Y2561 variant, right) estimated by MD simulations. The counts of MD models are shown as a heat map and as histograms along the tilt and twist axes. The heat map colors are defined by counts, as indicated on the right. Highly populated SD1/SD2 arrangements (clusters) are indicated with labels I (wt, Y2561), II (wt, Y2561), and III (Y2561 only). A scheme illustrating the definition of twist and tilt angles is inserted, using colors consistent in panel B. (B) Representative examples of SD1/SD2 arrangement clusters I, II, and III are shown, and their approximate population densities are indicated by “+” signs. Centers of mass used for calculating tilt and twist angles are shown as colored spheres and connected by gray sticks.
The separate C4 domain is capable of platelet integrin αIIbβ3 binding
The most pronounced function of the C4 domain, contrary to all other VWF C domains, is to bind platelet integrin αIIbβ3.1 To confirm that the C4 domain construct we used for structure determination and MD simulations is functional, we used a solid-phase binding assay to measure its interaction with αIIbβ3 (Figure 1B). The wt C4 domain binds to integrin αIIbβ3 with a half-maximal binding of 200 nM. As expected, C4 domain mutants, in which the 2 integrin binding residues of the RGD motif were substituted either by alanines or glutamates, lost the ability to bind integrin αIIbβ3.
The VWF Y2561 C4 domain variant has a similar structure and dynamics
Using the wt C4 domain (F2561) as reference, we set out to probe possible structural effects of the Y2561 variant. This VWF variant leads to a significant increase of myocardial infarction risk.15 The corresponding residue is located at the distal face of SD2, close to the C-terminus of the C4 domain, with its side chain presented on the surface (Figure 1C). The overall structure of this variant is not perturbed, as only minimal chemical shift differences of 1H-15N resonances can be observed (supplemental Figure 1), whereas the 15N relaxation profile also exhibits no significant changes (Figure 2A). Nevertheless, a slight increase of the global rotational correlation time can be observed (Figure 2A,C). This could be attributed to a higher oligomerization tendency for the Y2561 variant, which also explains the dependence of τc upon increasing concentrations. The change of F2561 to tyrosine also affects the Rex of V2547, indicated by altered magnetic field dependence (Figure 2C-D).
Using the same MD approach employed for the wt C4 domain, we show that the 2-microsecond ensembles of the Y2561 C4 variant are broadly similar: although we observed only minor changes in terms of SD1/SD2 tilt, we found a third minor population in the twist analysis peaked at φ ∼115°, denoted as cluster III (Y2561) (Figure 3A-B right). This cluster has a significantly bent SD1/SD2 arrangement similar to cluster II, which has been found in both the wt C4 and Y2561 C4 variant domains. Calculated relaxation profiles also confirmed that the mutation does not appear to affect the picosecond-nanosecond behavior of the 2 C4 subdomains (Figure 2B). Thus, aside from potential contributions from minor conformations likely associated with hidden conformational changes suggested by the Rex of V2547, we found no direct influence of the Y2561 variant on C4 dynamics itself. The Y2561 variant also binds to integrin αIIbβ3 similarly to wt C4 domain (Figure 1B), demonstrating that it does not impair the RGD platelet integrin binding site.
Discussion
In this work, we present the first structure of a VWC domain of the VWF protein, from which the name of this domain was derived.52 Key findings, different from previous structures, are its peculiar, in part noncanonical disulfide pattern, and the presence of an exposed and flexible loop region containing the RGD platelet integrin-binding motif, accessible for binding to integrin (supplemental Figure 2C). The structure of the C4 domain also indicates that the GOF Y2561 variant15 is unlikely to be associated with an alteration of platelet integrin binding. The structural position of this variant is close to the C-terminus of the C4 domain (Figure 1C), suggesting that instead it could play a role in the interface with the succeeding neighboring domain C5 thereby affecting shear-induced VWF self-association. Proof of such model will require additional structural data on larger VWF fragments covering both C4 and C5 domains.
To determine unique features of the VWF C4 domain in relation to other VWC domains, we compared the arrangement of its 2 subdomains SD1 and SD2 with those of known VWC structures: Collagen IIA (Col2a),22,23 CCN3,23 and CV-2.24,25 As the basis for structural comparison, we used a total of 24 residues (Figure 4A boxed segments) covering the 3 β-strands I-II-III of the second larger SD1 β-sheet for superimposition with that of the crystal structures of Col2a, CCN3, and CV-2. Only these 3 segments are sufficiently structurally conserved to provide a reliable structure-based sequence alignment (Figure 4A,C). There are 2 conserved cysteines within the second boxed segment that are both engaged in disulfide bridges, indicating a conserved role in VWC domain fold determination. In contrast, such structure-based alignment is not possible for SD2 due to the lack of sufficiently conserved structural elements and sequence.
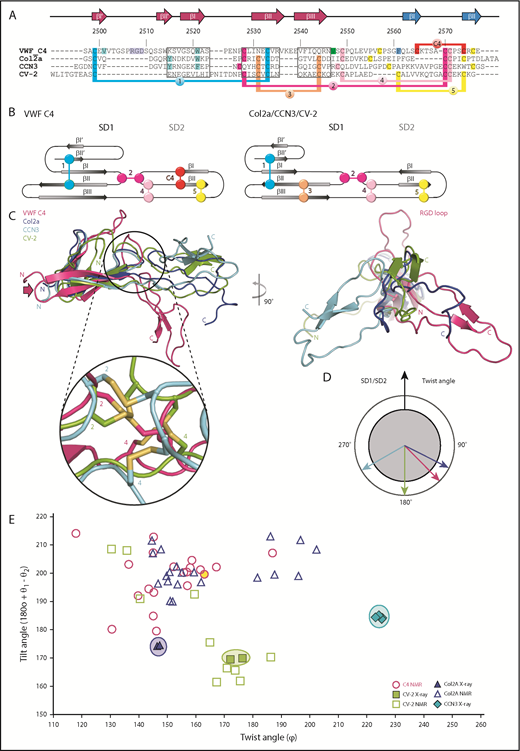
Structural comparison of the VWF C4 domain with other known VWC structures. (A) Sequence alignment of VWF C4, Col2a (PDB entry 5NIR), CCN3 (PDB 5NB8), and CV-2 (PDB 3NK3) with 3 stretches of structurally aligned residues using PDBeFold36 marked in boxes. The remaining residue stretches were manually aligned, using the conserved cysteine pattern as a guideline. The VWF C4 domain residue numbers and secondary structural elements are shown on top of the alignment. Disulfide bridges are highlighted in different colors, demonstrating the different levels of positional conservation among the different VWC sequences numbered as in Figure 1C and Table 2. VWF residues that are shown in the structure presentation of the C4 domain (Figure 1C) are highlighted in corresponding colors. (B) Schematic representations of distinct disulfide patterns in VWF C4 (left) and Col2a/CCN3/CV-2 (right), using the same colors as in panel A. (C) Superimposition of all 4 VWC domain structures aligned to SD1, showing a considerably more kinked SD1/SD2 arrangement in the lowest energy VWF C4 conformer (compare Figure 1C) than those observed for the other 3 VWF domain structures (left). Inset zooming into the SD1/SD2 interface, demonstrating structural diversity of the interdomain disulfide bridges 2 and 4. For visual clarity, the zoomed-in disulfide pattern of Col2a, which is closely related to that of CCN3, is not shown. When viewed along SD1, the twist angle of CCN3 is the most distinct from those observed for VWF C4, Col2a, and CV-2 (right). (D) Schematic representation of twist angles calculated for representative VWC domain structures (panel C, right). (E) Twist/tilt angle analysis of VWC structures. Angle clusters of crystal structures with multiple copies are highlighted with circles in corresponding colors. The lowest energy conformation of the VWF C4 domain used in other figures is highlighted in yellow.
Structural comparison of the VWF C4 domain with other known VWC structures. (A) Sequence alignment of VWF C4, Col2a (PDB entry 5NIR), CCN3 (PDB 5NB8), and CV-2 (PDB 3NK3) with 3 stretches of structurally aligned residues using PDBeFold36 marked in boxes. The remaining residue stretches were manually aligned, using the conserved cysteine pattern as a guideline. The VWF C4 domain residue numbers and secondary structural elements are shown on top of the alignment. Disulfide bridges are highlighted in different colors, demonstrating the different levels of positional conservation among the different VWC sequences numbered as in Figure 1C and Table 2. VWF residues that are shown in the structure presentation of the C4 domain (Figure 1C) are highlighted in corresponding colors. (B) Schematic representations of distinct disulfide patterns in VWF C4 (left) and Col2a/CCN3/CV-2 (right), using the same colors as in panel A. (C) Superimposition of all 4 VWC domain structures aligned to SD1, showing a considerably more kinked SD1/SD2 arrangement in the lowest energy VWF C4 conformer (compare Figure 1C) than those observed for the other 3 VWF domain structures (left). Inset zooming into the SD1/SD2 interface, demonstrating structural diversity of the interdomain disulfide bridges 2 and 4. For visual clarity, the zoomed-in disulfide pattern of Col2a, which is closely related to that of CCN3, is not shown. When viewed along SD1, the twist angle of CCN3 is the most distinct from those observed for VWF C4, Col2a, and CV-2 (right). (D) Schematic representation of twist angles calculated for representative VWC domain structures (panel C, right). (E) Twist/tilt angle analysis of VWC structures. Angle clusters of crystal structures with multiple copies are highlighted with circles in corresponding colors. The lowest energy conformation of the VWF C4 domain used in other figures is highlighted in yellow.
The long hairpin loop comprising the RGD platelet integrin-binding motif is unique in the C4 domain (Figure 4A,C right). In contrast, the β-strands of the first sheet I′-II′ in Col2a and CCN3 are each connected by a short 2-residue hairpin only. The difference between the C4 domain and the structures of Col2a and CCN3 is further reflected by how the 2 SD1 β-sheets are packed with each other. While in Col2a and CCN3, there is a π/π-interaction from 2 aromatic residues in the 2 SD1 β-sheets23 ; in C4, the equivalent interaction is formed by V2501 (βI′) and W2521 (βI) (Figure 1C; turquoise highlights in Figure 4A). We note that in the CV-2 structure, the N-terminal part of SD1 is unrelated and partially unfolded.
A key signature of the VWC domains lies within their partially conserved pattern of disulfide bridges (Figure 4A-B; Table 2). Although the first of the 2 canonical intra-SD1 disulfide bridges is present in all 4 known VWC structures, the second one (number 3) is missing in the VWF C4 domain. Interestingly, the 2 β-strands without the canonical disulfide bridge are connected by an extended, charged loop in the VWC C4 domain (residues 2536-2539, indicated by an arrow in Figures 1C and 4C) that is missing in the other 3 structures of Col2a, CCN3, and CV-2. We notice that this loop is on the opposite side of the C4 platelet integrin-binding site (Figure 1C). Considering a zipped stem arrangement of the C-terminal VWF segment as inferred from available low-resolution data10 (Figure 1A), this loop may be in an attractive position to contribute to zipped VWC dimerization. Along with the additional intra-SD2 disulfide bond (number C4), these unique structural features in the C4 domain may impact the conformational stability of both SD1 and SD2, which merit further investigation in expanded structural studies.
The 2 SD1/SD2 connecting disulfide bridges (numbers 2 and 4), although being in related sequence positions in the structure-based alignment, are in structurally quite divergent positions in the C4 domain when compared with the other VWC structures (Figure 4C zoomed-in inset). Taking the data together, our findings indicate that limited sequence and structural variability of an otherwise conserved disulfide pattern are main determinants of SD1/SD2 arrangement differences in the VWC structures. In the VWF C4 domain the focal point of this hinge can be localized to residue V2547, which is close to the disulfide bridge number 4 (Figure 1C). To determine whether this observation is part of a general mechanism, it would be interesting to confirm the existence of similar focal SD1/SD2 hinge residues in the related VWC structures by measuring their dynamics as well.
For a more quantitative comparison, equivalent SD1/SD2 hinge and complementary angles were computed the same way as for the C4 MD analysis (Figure 3), including the deposited lowest-energy NMR ensembles of Col2a and CV-2 (Figure 4E). In the absence of a subsequent MD analysis available for those NMR structures, we limit our further comparison with the 3 known crystal structures, keeping in mind that their arrangements may be artificially rigidified due to crystal lattices. Although the CCN3 crystal structure displays a twist angle of ∼225° leading to a right-handed kinked SD1/SD2 arrangement, the Col2a structures reveal an opposite left-handed bent with a twist angle of <150°. In contrast, the SD1/SD2 arrangement of CV-2 is almost entirely extended with a twist angle of close to 180°. Naturally, the angles observed in the different NMR ensembles are more diverse (Figure 4E), as long distances between SD1 and SD2 are less defined.
In our MD simulations, an extended SD1/SD2 arrangement of the C4 domain is most populated, with twist angles deviating <10° from a 180° angle (region I in Figure 3A-B). The presence of neighboring VWC domains (C1-C3 and C5-C6) may indeed impact the population of different SD1/SD2 arrangements, and hence, dimeric stem formation and platelet integrin accessibility.15
Ultimately, high-resolution structures of multidomain stretches of the VWF C-segment are needed for further elucidation of the molecular requirements for parallel dimerization, integrin binding, and the effect of external shear forces in these processes. Such studies could also unravel further insights into previously established differences in the predicted disulfide pattern of odd- (1, 3, 5) and even- (2, 4, 6) numbered VWF C domains,3 and how they arrange in possible tandems (1/2, 3/4, 5/6) with very short linkers in between.
The structure of VWF C4 has been deposited in the Protein Data Bank under accession code 6FWN. The NMR assignments for VWF C4 have been deposited in the Biological Magnetic Resonance Data Bank under accession code 34243.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors are grateful to Kai Horny, who assisted in calculations, and thank Remco Sprangers for sharing the CPMG pulse sequence.
E.-R.X., K.K., C.A.-S., F.G., R.S., and M.W. acknowledge the SHENC research grant (FOR1543) from the Deutsche Forschungsgemeinschaft. P.-C.C. is supported via the EI3POD program by EMBL and the EU Marie Curie Action Cofund grant 664726.
Authorship
Contribution: E.-R.X. conceived the study, produced the protein, analyzed the structure, and wrote the manuscript; S.v.B. and J.H. performed the NMR experiments, structure calculation, and dynamics analysis; P.-C.C. conducted MD simulations, NMR spin-relaxation computations, and contributed to manuscript writing; P.J.L. and C.V.D. performed solid-phase binding assays; K.K., C.A.-S., and F.G. conducted MD simulations and dihedral angles analysis; B.S. optimized and set up the CPMG pulse experiment and did water refinement; J.F. conducted HSQC experiments; T.O. provided the VWF gene; F.G., R.S., and M.W. initiated the study; and M.W. and J.H. designed and supported the study and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Matthias Wilmanns, European Molecular Biology Laboratory, Hamburg Unit, Notkestrß 85, 22607 Hamburg, Germany; e-mail: matthias.wilmanns@embl-hamburg.de; and Janosch Hennig, European Molecular Biology Laboratory, Meyerhofstrß 1, 69117 Heidelberg, Germany; e-mail: janosch.hennig@embl.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal