

Key Points
D3 assembles by packing C8-3, TIL3, and E3 modules against the VWD module into a wedge from which TIL′ and E′ modules in D′ project.
Burial of cysteines protects them from dimerization in the ER and requires structural rearrangement for D′D3 dimerization in the Golgi.

Abstract
D assemblies make up half of the von Willebrand factor (VWF), yet are of unknown structure. D1 and D2 in the prodomain and D′D3 in mature VWF at Golgi pH form helical VWF tubules in Weibel Palade bodies and template dimerization of D3 through disulfides to form ultralong VWF concatemers. D′D3 forms the binding site for factor VIII. The crystal structure of monomeric D′D3 with cysteine residues required for dimerization mutated to alanine was determined at an endoplasmic reticulum (ER)-like pH. The smaller C8-3, TIL3 (trypsin inhibitor-like 3), and E3 modules pack through specific interfaces as they wind around the larger, N-terminal, Ca2+-binding von Willebrand D domain (VWD) 3 module to form a wedge shape. D′ with its TIL′ and E′ modules projects away from D3. The 2 mutated cysteines implicated in D3 dimerization are buried, providing a mechanism for protecting them against premature disulfide linkage in the ER, where intrachain disulfide linkages are formed. D3 dimerization requires co-association with D1 and D2, Ca2+, and Golgi-like acidic pH. Associated structural rearrangements in the C8-3 and TIL3 modules are required to expose cysteine residues for disulfide linkage. Our structure provides insight into many von Willebrand disease mutations, including those that diminish factor VIII binding, which suggest that factor VIII binds not only to the N-terminal TIL′ domain of D′ distal from D3 but also extends across 1 side of D3. The organizing principle for the D3 assembly has implications for other D assemblies and the construction of higher-order, disulfide-linked assemblies in the Golgi in both VWF and mucins.
Introduction
Von Willebrand factor (VWF) is an ultralarge, multifunctional glycoprotein with a key role in arteriolar hemostasis. The long length of VWF concatemers enables hydrodynamic flow to elongate VWF and exert mechanical tension that activates binding of VWF to platelets, as well as formation of the platelet plug that stanches arteriolar bleeding.1,2 Subsequently, the coagulation cascade stabilizes the plug by incorporation of fibrin.
The D′D3 assembly in VWF both enables multimerization and provides a binding site for coagulation factor VIII (FVIII).3-6 VWF is unusually cysteine-rich. Intrachain disulfide bonds and interchain disulfide bonds that dimerize VWF through its C-terminal cystine knot domain are formed in the endoplasmic reticulum (ER). In the Golgi and nascent Weibel-Palade bodies, the D1 and D2 assemblies of the VWF prodomain and D′D3 of mature VWF form helical tubules in a process that requires low pH and Ca2+.7,8 Disulfide linkages then form between D3 assemblies of VWF dimers that are adjacent in tubules. Ultralong VWF concatemers are thus formed in which VWF monomers are disulfide-linked tail to tail through their C-terminal cystine knot domains and head to head through their N-terminal D3 assemblies. During secretion, the pH of Weibel Palade bodies increases to the slightly alkaline pH of plasma, the prodomain dissociates, and unfurling of tubules releases ultralong VWF into the bloodstream.1
VWF binding to FVIII in the bloodstream extends FVIII half-life from 18 minutes in mice and fewer than 3 hours in humans to 6 to 8 hours in mice and 12 to 14 hours in humans.9-11 While greatly extending FVIII half-life, VWF ironically also imposes a ceiling on FVIII half-life extension of ∼16 hours, which resembles that of VWF itself. The frequency of FVIII deficiency (hemophilia A, 1 in 5000 males) and its morbidity, together with short VWF and FVIII half-life, require replacement therapy every 2 days and impose enormous burdens on patients and costs on healthcare systems. Therefore, the structure of VWF D′D3 is of immense medical importance for engineering extended half-life FVIII therapy.11 Moreover, mutations in D′D3 cause 4 different types of von Willebrand disease (VW disease), further demonstrating the medical importance of understanding D′D3 structure.
Here, we report a structure of monomeric D′D3 that addresses its interesting biological functions and defines the organizing principles of D assemblies. Four D assemblies are present in VWF (D1, D2, D3, and D4), which each contain 4 modular domains, von Willebrand D domain (VWD), C8 (8 cysteines), TIL (trypsin inhibitor-like), and E (in analogy to the A, C, and D repeats in VWF). D assemblies are so named because they appear in electron microscopy not as single globules similar to typical domains, but as compact assemblies of multiple lobules, in agreement with their content of 4 types of modules.12,13 Our structure shows how these modules pack against one another in a highly specific circular assembly that contrasts with the more typical extended orientation found between tandem domains in extracellular proteins and in D′. D′ contains only 2 modules, TIL′ and E′, and has been seen by nuclear magnetic resonance (NMR).3 The VWD and C8 modules in VWF have not previously been structurally characterized.
Cys1099 and Cys1142 are the only free cysteines in D′D3 monomers and have been suggested to form the interdimer disulfide bonds within VWF.14 A major unsolved question is how these 2 cysteines remain free in the ER. Premature interchain disulfide linkage in the ER would prevent assembly into tubules in the Golgi, and formation of ultralong VWF concatemers. Our crystal structure of monomeric D′D3 shows that the mutant C1099A and C1142A residues are not surface exposed, which provides a mechanism to prevent premature interchain disulfide formation. Finally, our structure provides insight into how mutations cause VW disease and defines the binding site within VWF for FVIII.
Methods
D1D2D′D3 with C1099A and C1142A mutations was expressed as an Fc-fusion protein with an intervening thrombin cleavage site (supplemental Figure 1, available on the Blood Web site; plasmid pSYN-VWF-039, deposited at AddGene) in HEK293F cells by transient co-transfection with vectors expressing D1D2D′D3-Fc and furin (92.5:7.5 mol:mole).5 To obtain high mannose glycans, 1 mg/mL kifunensine (GlycoSyn, Wellington, New Zealand) was added 4 hours after transfection. Purification with MabSelect SuRe (Sigma Aldrich) was as described,5 except washing was with phosphate-buffered saline containing 0.9 M NaCl and elution was with 150 mM NaCl, 25 mM sodium citrate at pH 3.4. Peak fractions were pooled and loaded onto a HiTrap DEAE Sepharose FF column (GE, Piscataway, NJ) equilibrated in 20 mM HEPES at pH 7.2 and eluted with a NaCl gradient. D′D3-Fc was exchanged into 150 mM NaCl, 20 mM HEPES at pH 7.2, and Fc was removed using Thrombin CleanCleave (Sigma Aldrich, Saint Louis, MO). Cleaved Fc was removed using MabSelect SuRe. Digested D′D3 was treated with 5 mM diisopropylfluorophosphate to inhibit residual thrombin. N-glycans were shaved with Endo Hf (New England Biolabs, Ipswich, MA), followed by removal of the EndoHf with amylose beads.
Purified D′D3 (supplemental Figure 2) was dialyzed into 150 mM NaCl, 20 mM HEPES at pH 7.4, and concentrated to 8 mg/mL. D′D3 was crystallized by vapor diffusion at 24°C with an equal volume of 5.5% (wt/vol) PEG8000, 10% (wt/vol) PEG1000, 0.2 M calcium acetate, and 0.1 M Tris⋅acetate at pH 8.5. Crystals grew to 250 µm × 100 µm × 100 µm) in 2 weeks. A platinum (Pt) derivative was prepared by soaking crystals for 5 minutes in reservoir solution containing 5 mM K2PtCl4. Crystals were briefly cryosoaked in reservoir solution containing 15% xylitol and flash-frozen in liquid nitrogen. Diffraction data were collected at the LRL-Collaborative Access Team beam line of Advanced Photon Source at −173°C, using a MAR225 charge coupled device detector.
Anomalous signal from the peak wavelength of Pt was used to determine the Pt substructure. Data were processed using the XDS program package. Friedel pairs were kept unmerged. PHENIX.AUTOSOL used HySS to identify heavy atom locations, PHASER to calculate phases using all reflections, and RESOLVE for density modification.15 The top 15 peaks were chosen as the starting Pt sites. Initial phases were extended to the 2.5-Å native data set. After density modification using RESOLVE, continuous density of secondary structure elements could be observed. AUTOBUILD was used to build one-third of the sequence, and was used several times in the first few of many iterative steps of manual building in COOT,16 refinement with PHENIX, and validation with MolProbity.17
Structure data sets were deposited at the Protein Data Bank with code 6N29. Diffraction images were deposited to the SBGrid Data Bank: Native, doi: 10.15785/SBGRID/628; Pt anomalous, doi: 10.15785/SBGRID/629; and sulfur anomalous, doi: 10.15785/SBGRID/630
Results
Overall structure of the D′D3 monomer
A D′D3 C1099A/C1142A crystal structure refined to 2.5 Å was solved by anomalous dispersion (supplemental Table 1; Figure 1A-B). Two nearly identical D′D3 molecules (A and B) are present in the asymmetric unit. Molecule A extends from residue 764, the first residue of mature VWF immediately after the furin cleavage site, to residue 1241 within the flexible, O-glycosylated segment between E3 and A113 (Figure 2). Electron density also reveals N-acetylglucosamine residues at each of 3 N-glycosylation sites previously determined by glycoprotein chemistry, including one in a noncanonical Asn1147-Ser-Cys sequence18 (Figure 1A-B). All 25 disulfide bonds within the D′D3 monomer are unambiguously assigned from composite omit simulated annealing electron density and anomalous signal from sulfur (supplemental Figure 3). Of 25 disulfides, 11 confirm chemical assignment,19 6 confirm homology assignment,13 5 confirm both homology assignment and NMR assignment,3 and 3 in C8-3 are newly assigned; 4 disulfides assigned chemically are contradicted (supplemental Figure 4).

Structure of D′D3 assembly. (A-B) Two views of D′D3 showing a rectangular face (A) and triangular face (B) of the D3 wedge with dimensions. (C) Interactions that stabilize the E′-VWD3 junction. (D) View of the face of D3 slightly below which cysteines implicated in D3 dimerization, mutated to alanine here, are buried. Figures prepared with PyMol show ribbon cartoon and disulfides and N-glycans with white carbons in stick, red oxygens, blue nitrogens, and yellow sulfurs. Important residues are shown in stick and labeled. Ca2+ is shown as a silver sphere. In panels A and B, the Cβ atoms of the C1099A and C1142A mutants are shown as spheres. In D and subsequent figures, A1099 and A1142 are modeled as cysteines, using the allowed rotameric position of the S atom, as discussed in Results. Dot surfaces around the S atom emphasize that there is room for it in the structure, and that it is a model.
Structure of D′D3 assembly. (A-B) Two views of D′D3 showing a rectangular face (A) and triangular face (B) of the D3 wedge with dimensions. (C) Interactions that stabilize the E′-VWD3 junction. (D) View of the face of D3 slightly below which cysteines implicated in D3 dimerization, mutated to alanine here, are buried. Figures prepared with PyMol show ribbon cartoon and disulfides and N-glycans with white carbons in stick, red oxygens, blue nitrogens, and yellow sulfurs. Important residues are shown in stick and labeled. Ca2+ is shown as a silver sphere. In panels A and B, the Cβ atoms of the C1099A and C1142A mutants are shown as spheres. In D and subsequent figures, A1099 and A1142 are modeled as cysteines, using the allowed rotameric position of the S atom, as discussed in Results. Dot surfaces around the S atom emphasize that there is room for it in the structure, and that it is a model.
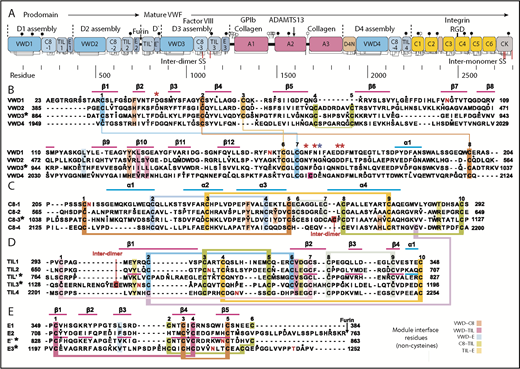
Domain architecture of VWF and sequence of modules in D assemblies. (A) Domain architecture. Cysteines are vertical lines and are connected for disulfide bonds. N- and O-linked glycans are closed and open lollipops, respectively. Domains are scaled to length, and residues are shown with pre-pro numbering. (B-E) D assembly modules are aligned by sequence with insertions and deletions moved to loops between secondary structures defined here; in addition, the TIL′ and TIL3 modules and E′ and E3 modules are aligned by structure. Modules in the crystal structure are indicated with an asterisk. Disulfide-linked cysteines defined in the structure and the newly predicted disulfide between C8-4 and TIL4 are linked with colored lines. Module interface residues are highlighted as shown in the key in the lower right. α helices and β strands are shown in cyan and magenta lines, respectively, above sequence blocks for the D3 assembly or above TIL′ and E′ sequences for D′. Glycosylated Asn residues are shown in red. Ca2+-coordinating residues are asterisked in red (sidechain) or blue (backbone). Dots appear above decadal residues.
Domain architecture of VWF and sequence of modules in D assemblies. (A) Domain architecture. Cysteines are vertical lines and are connected for disulfide bonds. N- and O-linked glycans are closed and open lollipops, respectively. Domains are scaled to length, and residues are shown with pre-pro numbering. (B-E) D assembly modules are aligned by sequence with insertions and deletions moved to loops between secondary structures defined here; in addition, the TIL′ and TIL3 modules and E′ and E3 modules are aligned by structure. Modules in the crystal structure are indicated with an asterisk. Disulfide-linked cysteines defined in the structure and the newly predicted disulfide between C8-4 and TIL4 are linked with colored lines. Module interface residues are highlighted as shown in the key in the lower right. α helices and β strands are shown in cyan and magenta lines, respectively, above sequence blocks for the D3 assembly or above TIL′ and E′ sequences for D′. Glycosylated Asn residues are shown in red. Ca2+-coordinating residues are asterisked in red (sidechain) or blue (backbone). Dots appear above decadal residues.
The C8-3, TIL3, and E3 modules wrap around the periphery of the larger VWD3 module to form the wedge-shaped D3 assembly. Figure 1A-B views D3 on rectangular and triangular faces of the wedge, respectively.
In D′, the TIL′ and E′ modules interact end to end to extend 6 nm from the D3 assembly (Figure 1B). The long axis of D′ projects at an angle of 60° from the rectangular wedge face. E′ connects to VWD3 near the middle of 1 rectangular face of the wedge, and near the middle of the interface of VWD3 with C8-3 (Figure 1B-C). The FVIII-binding end of TIL′ is thus positioned far from VWD3, optimizing its accessibility. D′ corresponds to the horn-shaped protrusion from D3 observed in previous electron microscopy studies of D′D3.4,5,13
Position of the C1099A and C1142A mutations
Intriguingly, the 2 cysteine residues implicated in D′D3 dimerization and mutated to alanine here are buried in hydrophobic pockets, yet lie near the solvent accessible surface. We tested whether there was space within the pockets to accommodate the additional sulfhydryl group of cysteine compared with alanine in 1 of 3 geometrically optimal sidechain rotamer orientations. Indeed, there is, as shown by the dot surfaces surrounding the sulfur atoms of the cysteine models (Figure 1D). This result suggests that the C1099A/C1142A mutations have not caused structural artifacts, and that our structure represents a native conformation of monomeric D′D3, consistent with crystallization at an ER-like alkaline pH. C1099A lies in the first helical turn of the C8-3 α4 helix at the interface with VWD3. It is buried by sidechains shown in stick in Figure 1D, including Met-872 and Val-919 of VWD3. In TIL3, the bulky sidechains of residues Leu-1135, Tyr-1140, and Trp-1144 participate in the burial of C1142 (Figure 1D). These results suggest that Cys-1099 and Cys-1142 are protected from dimerization in the ER, and that structural rearrangements are required for D3 dimerization in the Golgi (“Discussion”).
Module structures
Figure 3 shows each D′D3 module rainbow-colored to trace the path the polypeptide takes to create the fold, from N terminus (blue) to C terminus (red). VWD3 contains an unusually large β sandwich with 12 β strands (Figure 3A). The 9-10-11-12-1-2-3 β-sheet is built beginning with the β1 strand near the middle of the β-sheet. After β3, the polypeptide crosses over to form the 4-5-6-7-8 β sheet and then crosses again to form β strands 9 to 12. β12 is followed by a long meandering loop, the α1 helix, and the C-terminal loop, which pack over the β1, β2, and β3 strands (Figure 3A). VWD3 is stabilized by 3 long-range disulfide bonds that tie the β12-α1 loop to the β1 strand and β4-β5 loop, and the C-terminal loop to the β3-β4 loop. Unexpectedly, a Ca2+ ion binds to VWD3 and makes long-range connections between the β12-α1 loop and the β2-β3 loop (Figure 3A-B; supplemental Figure 3). Ca2+ coordination by Asp and Asn sidechains is stabilized by an extensive surrounding hydrogen bond network (Figure 3B). VWD3 is structurally homologous to the C-terminal domain of repulsive guidance molecules20 (supplemental Figure 5), yet has only 10% sequence identity. In contrast to VWD3, the repulsive guidance molecule VWD domain does not associate with other modules to form an assembly, and lacks a Ca2+-binding site.
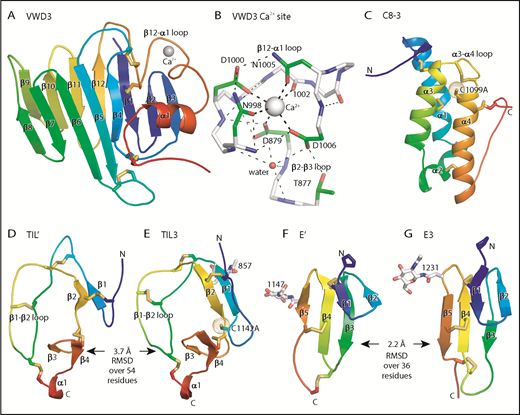
Structure of D assembly modules. (A,C-G) Each module is shown rainbow colored to trace the path the polypeptide takes to create the fold, from N terminus (blue) to C terminus (red). (B) The Ca2+-binding site. Mainchain carbons are shown in white, with sidechain carbons in green. Ca2+ and a water oxygen are shown as spheres. Coordination bonds to the Ca2+ and hydrogen bonds are shown as thick and thin dashed lines, respectively. Other representations are as in Figure 1. Superposition of TIL domains required the Deep Align server27 ; E modules were superimposed with PyMol. Superimposed structures are shown separated horizontally on the page. Structural differences between modules are shown as root mean square deviation of Cα atoms.
Structure of D assembly modules. (A,C-G) Each module is shown rainbow colored to trace the path the polypeptide takes to create the fold, from N terminus (blue) to C terminus (red). (B) The Ca2+-binding site. Mainchain carbons are shown in white, with sidechain carbons in green. Ca2+ and a water oxygen are shown as spheres. Coordination bonds to the Ca2+ and hydrogen bonds are shown as thick and thin dashed lines, respectively. Other representations are as in Figure 1. Superposition of TIL domains required the Deep Align server27 ; E modules were superimposed with PyMol. Superimposed structures are shown separated horizontally on the page. Structural differences between modules are shown as root mean square deviation of Cα atoms.
The previously structurally uncharacterized C8 domain is defined by sequence homology (Pfam PF08742). The VWF C8-3 module consists of a bundle of 3 helices, α1, α3, and α4, and a shorter α2 helix (Figure 3C). Four disulfides in C8-3 shared with all C8 domains in VWF (Figure 2C) stabilize interactions of α-helices with other α-helices or with the N- and C-terminal loops (Figure 3C). A fifth disulfide in the α3-α4 loop is present in only some C8 domains. Nearby, in the first helical turn of α4, is Cys-1099 (Figure 3C), which is required for D′D3 dimerization.14 Cys-1099 locates to a 2-residue insertion in C8-3 compared with the C8-1 and C8-2 domains (Figure 2C).
Although TIL′ and TIL3 each have the trypsin inhibitor-like fold and 5 disulfide bonds, the length of their β1 and β2 strands and conformation of their β1-β2 loops differ, resulting in a large Cα atom RMSD of 3.7 Å (Figure 3D-E). The TIL′ crystal structure and the lowest-energy TIL′ NMR structure have a Cα atom root mean square deviation (RMSD) of 3.5 Å, in agreement with marked dynamics of the TIL′ N-terminal and β1-β2 loops (Figure 4A-C).3 Among TIL domains in VWF, TIL3 uniquely contains a 10-residue insertion containing Cys-1142 that functions in D′D3 dimerization (Figure 2D).14 This insertion markedly increases the length of the first loop and β1 strand in TIL3 compared with TIL′ (Figure 3D-E).
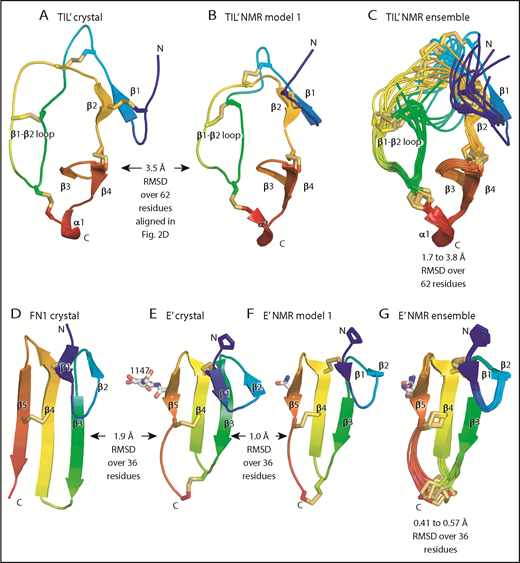
Comparisons of TIL and E modules. (A-G) Each module is shown rainbow-colored to trace the path the polypeptide takes to create the fold, from N terminus (blue) to C terminus (red). Structural differences between modules are shown as RMSD of Cα atoms. Within NMR ensembles, the RMSD ranges are between model 1 and models 2 to 10. Other representations are as in Figure 1. Superposition was with the Deep Align server.27 Protein Data Bank ID codes are 2MHP (D′ NMR structure) and 3MQL (sixth fibronectin type I domain of fibronectin).
Comparisons of TIL and E modules. (A-G) Each module is shown rainbow-colored to trace the path the polypeptide takes to create the fold, from N terminus (blue) to C terminus (red). Structural differences between modules are shown as RMSD of Cα atoms. Within NMR ensembles, the RMSD ranges are between model 1 and models 2 to 10. Other representations are as in Figure 1. Superposition was with the Deep Align server.27 Protein Data Bank ID codes are 2MHP (D′ NMR structure) and 3MQL (sixth fibronectin type I domain of fibronectin).
E′ and E3 are small β sandwich domains with a 1-2 β sheet that packs against the 3-4-5 β sheet3 (Figure 3F-G). As predicted,13 E modules structurally resemble and share 2 disulfide bonds with fibronectin type I modules and the first tandem domain in VWC modules3 (Figure 4D-E).
Interaction between modules
The importance of intermodule interfaces was examined by quantitating the solvent-accessible surface area buried between them. Similarly, the stability of module orientation was measured as the difference in orientation between successive domains between molecules A and B in the crystal structure, and for D′ in the NMR structure. The orientation between TIL′ and E′ differed by 7° to 13° (Table 1). Although the area buried between these tandem domains is small, the CPC motif at their junction containing the last disulfide-bonded Cys of TIL′, a Pro residue, and the first disulfide-bonded Cys of E′, constrains rotation. Less rotation was found at the interface between E′ and VWD3, which is the smallest in D′D3 (Table 1), yet is stabilized by a disulfide bond, van der Waals contact of Val-862 in E′ with Gln-989 and Phe-999 in VWD3, and extensive flanking hydrogen bond networks (Figure 1C).
Buried solvent accessible interface between modules (Å2) and difference in intermodule angle between molecules A and B and for D′ the NMR structure (°)
| Interface | Buried solvent-accessible surface area, Å2 | Angular difference, ° |
|---|---|---|
| TIL`–E` | 540 | 7, 11, 13 |
| E`–VWD3 | 200 | 4.4 |
| VWD3–C8-3 | 1290 | 0.7 |
| VWD3–TIL3 | 1080 | NA |
| VWD3–E3 | 550 | NA |
| C8-3–TIL3 | 650 | 0.1 |
| TIL3–E3 | 430 | 0.6 |
| Interface | Buried solvent-accessible surface area, Å2 | Angular difference, ° |
|---|---|---|
| TIL`–E` | 540 | 7, 11, 13 |
| E`–VWD3 | 200 | 4.4 |
| VWD3–C8-3 | 1290 | 0.7 |
| VWD3–TIL3 | 1080 | NA |
| VWD3–E3 | 550 | NA |
| C8-3–TIL3 | 650 | 0.1 |
| TIL3–E3 | 430 | 0.6 |
NA, not applicable.
In contrast to D′, the large size of the intermodule interfaces in D3, and the small differences between molecules A and B in the angle between successive modules (Table 1), strongly support the hypothesis that D assemblies are created by highly specific associations between their modules. The interactions of VWD3 with C8-3 and with TIL3 are particularly large and have a significant hydrophobic component (Table 1; Figure 5). In Figure 5, the sidechains of all residues in D3 that make contacts (within 3.7 Å) of the backbone or sidechain of a different module (excluding residues in junctions between modules) are shown, all intermodule hydrogen bonds are shown as dashed lines, and residues that may make particularly significant contacts are labeled. All disulfide bridges are also shown. Histidine residues change protonation between alkaline ER pH and acidic Golgi pH and are overrepresented in module interfaces (Figure 5), comprising 5 (10.4%) of 48 interface residues compared with 3.2% overall in D3.
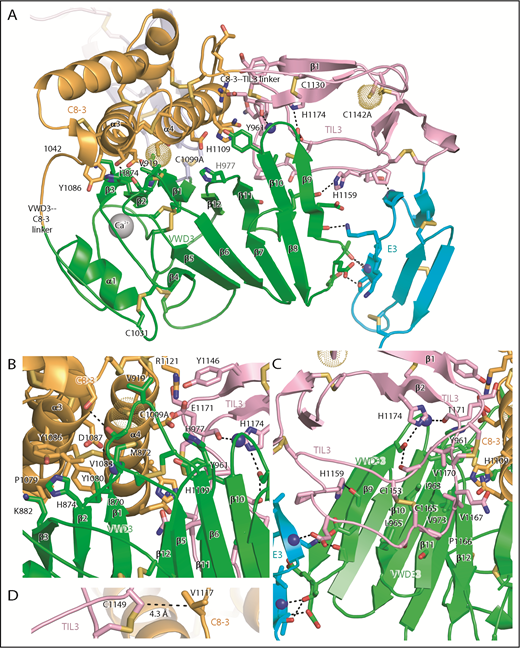
Intermodule interfaces in D3. (A) Overview. (B) View down the α3 and α4 helices of C8-3, emphasizing interfaces with VWD3 and TIL3. (C) View of TIL3 interfaces, with the view rotated ∼180° about the axis vertical in the page relative to A to show the opposite side of β strands 9 to 12. All residues that interact across module interfaces (with heavy atoms within 3.7 Å, excluding at intermodule peptide connections) are shown with sidechains and hydrogen-bonding backbone carbonyl groups in stick and nitrogens as spheres. Intermodule hydrogen bonds are shown as dashed lines. (D) Residues in D3 that align with cysteines in D4 and that have Cβ atom distances (dashed line) ideal for disulfide bond formation in D4 between the TIL4 and C8-4 modules. Important residues are labeled. Other representations are as in Figure 1.
Intermodule interfaces in D3. (A) Overview. (B) View down the α3 and α4 helices of C8-3, emphasizing interfaces with VWD3 and TIL3. (C) View of TIL3 interfaces, with the view rotated ∼180° about the axis vertical in the page relative to A to show the opposite side of β strands 9 to 12. All residues that interact across module interfaces (with heavy atoms within 3.7 Å, excluding at intermodule peptide connections) are shown with sidechains and hydrogen-bonding backbone carbonyl groups in stick and nitrogens as spheres. Intermodule hydrogen bonds are shown as dashed lines. (D) Residues in D3 that align with cysteines in D4 and that have Cβ atom distances (dashed line) ideal for disulfide bond formation in D4 between the TIL4 and C8-4 modules. Important residues are labeled. Other representations are as in Figure 1.
The C8-3, TIL3, and E3 modules wrap around VWD3 on its face formed by the broad 9-10-11-12-1-2-3 β sheet, whereas the 4-5-6-7-8 β sheet and α1 helix have no interaction and are solvent exposed (Figure 5A). The hairpin loops that join adjacent β strands in each of these sheets form 2 long edges of the β sandwich. The edge facing the viewer in Figure 5A contains the β9-β10, β11-β12, and β1-β2 hairpin loops, which contain residues that interact with the C8-3 and TIL3 modules. The interaction with C8-3 is centered at the junction between these hairpin loops and their connecting β strands. In addition, the β5-β6 loop forms a long projection from the 4-5-6-7-8 β sheet to interact with C8-3. Val-919 in the β5-β6 loop partially buries C1099A (Figure 5A-B). As TIL3 wraps further around VWD3, the interaction moves across the 9-10-11-12-1-2-3 β sheet to the β11, β10, and β9 strands, and toward the middle of the β strands rather than their loop-bearing ends (Figure 5A-C). As wrapping is completed at E3, interaction shifts from the face of the 9-10-11-12-1-2-3 β sheet to its edge at the β9 strand, and further down the β sheet to include the β8-β9 loop (Figure 5A).
The C8-3 module interacts through its α3 and α4 helices with VWD3 (Figure 5B). Hydrophobic interacting residues on α3 include Pro-1079, Tyr-1080, Val-1083, and Tyr-1086. Interactions on VWD3 include hydrophobic residues Ile-870, Met-872, Val-919, and the aliphatic portion of Lys-882. The hydrogen bond of the Asp-1087 sidechain to the main chain of Met-872 is strengthened by its hydrophobic environment (Figure 5B). TIL3 interacts with its long β1-β2 loop with hydrophobic residues in the VWD3 β10 and β11 strands (Figure 5C). Interacting TIL3 residues include the C1153-C1165 disulfide, Pro-1166, Val-1167, and Val-1170. The sidechains of 2 TIL3 His residues, 1159 and 1174, hydrogen bond to VWD3 sidechain and backbone oxygen atoms. Moreover, the backbone of TIL3 residue 1171 hydrogen bonds to the backbone of the VWD3 β9-β10 loop (Figure 5C). E3 forms a smaller interface with VWD3 that includes multiple hydrogen bonds, but also a small interface at Thr-951 with VWD3 (Figure 5A,C) that is conserved as hydrophobic in other D assemblies (Figure 2B,E). Compared with the TIL′-E′ junction, the extra residue at the TIL3-E3 junction (Figure 2E) extends its β1 strand and causes E3 to swing toward VWD3 (Figures 3F-G and 6).
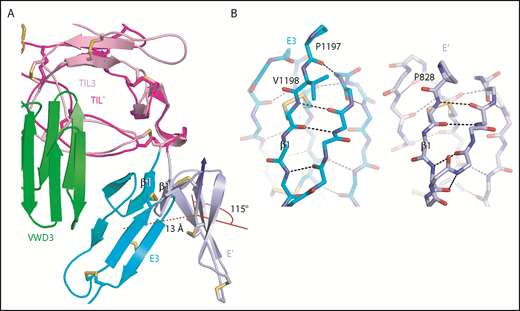
An extra residue in E3 alters TIL-E module orientation. E3 has 1 more N-terminal residue than the other E domains in VWF (Figure 2E). (A) Superposition on TIL3 of TIL′ shows how TIL3- E3 orientation brings E3 much closer to VWD3 than would occur with the TIL′-E′ orientation. There is a 115° difference in orientation angle about the axis shown with the cone and cylinder and a 13-Å difference in E module center of mass. (B) E3 and E′ are shown in same orientation after superposition and horizontal separation on the page. The extra N-terminal residue in E3 extends the β1-β2 β ladder by 1 position and the rotation of the polypeptide backbone alters the orientation of P1197 relative to P828 and results in the 115° rotation at the TIL-E3 interface, as shown in panel A.
An extra residue in E3 alters TIL-E module orientation. E3 has 1 more N-terminal residue than the other E domains in VWF (Figure 2E). (A) Superposition on TIL3 of TIL′ shows how TIL3- E3 orientation brings E3 much closer to VWD3 than would occur with the TIL′-E′ orientation. There is a 115° difference in orientation angle about the axis shown with the cone and cylinder and a 13-Å difference in E module center of mass. (B) E3 and E′ are shown in same orientation after superposition and horizontal separation on the page. The extra N-terminal residue in E3 extends the β1-β2 β ladder by 1 position and the rotation of the polypeptide backbone alters the orientation of P1197 relative to P828 and results in the 115° rotation at the TIL-E3 interface, as shown in panel A.
Discussion
D assembly structure and function
We have uncovered the organizing principles of D assemblies. The VWD3 module provides a large stable surface containing a broad β sheet against which the succeeding 3 modules pack. The long linkers between VWD and C8 and between C8 and TIL in D3 have few stabilizing interactions (Figures 1D and 5A), and are conserved in other D assemblies (Figure 2). These linkers are interesting because they would allow movement of C8 and TIL relative to VWD. Additionally, the α-helical structure of C8 and loose structure of TIL would permit localized structural change. In contrast, these modules are flanked at each end of the assembly by VWD and E with β sandwich structures that make them more rigid.
Our high-resolution structure of D3 provides an excellent model for understanding other D assemblies. The sequences of their modules are conserved, including the unexpected Ca2+-binding site in VWD (Figure 2). Whether this or another Ca2+-binding site is responsible for the Ca2+-dependence of VWF tubule formation7 remains to be determined. Key residues in module interaction interfaces in D3 that are conserved in D1, D2, and D4 (highlighted in Figure 2B-E) suggest that the orientation between modules described here for D3 is similar in other D assemblies. Our structure extends and confirms all previous disulfide predictions based on homology and loss or gain of pairs of cysteines in VWF,13 and enables another prediction. In D4, TIL4 lacks a Cys partner for Cys-1149 in TIL3, whereas C8-4 contains an extra Cys that aligns with Val-1117 in C8-3 (Figure 2C-D). Furthermore, the equivalent residues in D3, Cys-1149 and Val-1117, neighbor one another at distances ideal for disulfide bond formation (Figure 5D). These results suggest the presence of an intermodule disulfide bond between C8-4 and TIL4. Moreover, a triad of interacting residues at the C8-3–TIL-3 interface, Arg-1121, Tyr-1146, and Glu-1171 (Figure 5B), are conserved in other D assemblies (Figure 2C-D).
Thus, the module orientations found here in D3 appear highly conserved in other D assemblies, including in D1, D2, and D4 in VWF and in 6 homologous gel-forming mucins that, similar to VWF, have D1D2D′D3 architectures, form dimers through C-terminal cystine knot domains in the ER, and multimerize in the Golgi through D′D3.21 It is unusual for distinctive modules to be found in isolation from one another in some proteins, and in other proteins to occur in a specific order in the sequence and to organize into an assembly with a specific intermodule geometry. The superdomain organization of D assemblies may have evolved to accomplish 2 tasks. First, the large size of the D1, D2, and D3 assemblies of about 380 residues provides a large framework for formation of helical VWF tubules in Weibel-Palade bodies.7,8 Second, the putative flexibility and small size of the C8 and TIL modules of ∼90 and ∼60 residues, respectively, together with the unconstrained VWD-C8 and C8-TIL linkers, may facilitate structural rearrangements required for assembly into helical tubules and exposure of the cysteines that dimerize D′D3.
pH-dependent helical tubule formation and D3 dimerization
At the alkaline pH of the ER and the buffer used for crystallization here, histidine residues are largely unprotonated. Burial of residues 1099 and 1142 in our structure appears to be a biological mechanism to protect against aberrant, premature disulfide bond formation in the ER. On transport of VWF dimers to the trans-Golgi (pH 6.2) and Weibel-Palade bodies (pH 5.4), histidine residues (pKa ∼6) become protonated. Formation of disulfide-linked D′D3 dimers is dependent on the VWF prodomain, which is composed of the D1 and D2 assemblies.22 Furthermore, association of D1D2 and D′D3 into tubules is dependent on pH and Ca2+.7 Therefore, lower pH, Ca2+, and binding to D1D2 may be required to induce the structural changes in C8-3 and TIL3 that enable their cysteines to form interdimer disulfide bonds.
Each VWF tubule in a Weibel-Palade body appears to contain a single, ultralong VWF concatemer with VWF dimers linked through interdimer disulfides in D3 (Figure 2A).1 However, the cap monomers at each end should have unpaired D3 cysteines. After secretion into blood at pH 7.4 and dissociation of the prodomain, the D3 caps should adopt the structure crystallized here with buried free cysteines. Burial would prevent spurious disulfide formation between the free cysteines in the terminal D3 monomers and other plasma proteins.
His-874 in VWD3 forms a T-shaped aromatic interaction with Tyr-1086 in C8-3 that, on protonation of His, would convert to a stronger π-cation bond (Figure 5A). Although His-874 is not required for D′D3 dimerization, mutation of its equivalent in D2, His-395 (Figure 2B), strongly blocks pH-dependent dimerization.21 Tyr-1086 is conserved as Tyr-610 in D2 (Figure 2C), and thus a π-cation bond between His-395 and Tyr-610 in D2 may regulate pH-dependent D′D3 dimerization. Four other His residues required in D1 and D2 for D′D3 dimerization21 map to the periphery of the D assembly (Figure 7A) and suggest that this peripheral region in D assemblies mediates association among and organization into tubules of D1, D2, and D′D3. Mutation of His-1159 abolished VWF secretion,21 showing a critical role for this residue in the TIL3–VWD3 interface (Figure 5A).

Important histidines in D assemblies, specific VW type 2A and 2N disease mutations, and FVIII binding model. (A) Conserved histidines in D assemblies that when mutated, diminish or abolish D3 dimerization. Four histidines in D2, 1 in D1, and none in D3 abolish dimerization when mutated to alanine.21 These histidine residues are labeled next to the positions of the homologous residues in D3 which are shown as Cα atom spheres. (B) VW disease 2A and 2N mutations that are likely to be specific: 2A and 2N mutations that represent mutation of Cys or to Cys or are also reported as type 1 or 3 mutations have been excluded. Positions of mutations are shown as Cα atom spheres color-coded as in the key to right. Mutated residues are labeled. The distance from the centers of mutations in TIL′ and C8-3 is shown as a dashed line.
Important histidines in D assemblies, specific VW type 2A and 2N disease mutations, and FVIII binding model. (A) Conserved histidines in D assemblies that when mutated, diminish or abolish D3 dimerization. Four histidines in D2, 1 in D1, and none in D3 abolish dimerization when mutated to alanine.21 These histidine residues are labeled next to the positions of the homologous residues in D3 which are shown as Cα atom spheres. (B) VW disease 2A and 2N mutations that are likely to be specific: 2A and 2N mutations that represent mutation of Cys or to Cys or are also reported as type 1 or 3 mutations have been excluded. Positions of mutations are shown as Cα atom spheres color-coded as in the key to right. Mutated residues are labeled. The distance from the centers of mutations in TIL′ and C8-3 is shown as a dashed line.
VW disease mutations and binding to factor VIII
VW disease mutations are found throughout D′D3. Many mutations that result in partial (type 1) or complete (type 3) deficiency of VWF appear to disrupt module structure by altering packing in the hydrophobic core or removing or adding cysteines and altering disulfide bonds (supplemental Figure 6). Mutations at some residues, either to the same or a different amino acid, may be classified as causing more than 1 type of VW disease, including both qualitative type 2 and quantitative type 1 and 3 deficiencies.23 We therefore focus on mutations in type 2A and type 2N of residues at which type 1 or 3 mutations are not found, and are thus likely to have “specific” effects.
Type 2A mutations result in the absence of long VWF multimers. The S979N mutation in C8-3 introduces a putative N-glycosylation site into its interface with VWD3 in close proximity to Cys-1099 (Figure 5B). Thus, S979N may disrupt proper intermodule orientation or structure around Cys-1099 required for D3 dimerization.
Type 2N mutations cause defective binding to FVIII and map to D′ and D33 (Figure 7B). D879N in the VWD3 Ca2+-binding site disrupts the dual function of Asp-879 in coordinating Ca2+ and hydrogen bonding to the backbone of the Ca2+-binding loop (Figure 3B). Interestingly, D879N causes both type 2A and 2N VW disease,24 showing that the VWD3 Ca2+-binding site is required both for efficient D3 dimerization and binding of FVIII. The specific type 2N Q1053H and E1078K mutations lie in C8-3 and decrease FVIII binding to recombinant VWF by 10-fold and more than 10-fold, respectively.25,26 Recent negative stain electron microscopy studies on FVIII–D′D3 complexes suggest that the FVIII C1 domain interacts with D′ and appears to touch the D3 platform.4,5 Our results further suggest that there are 2 noncontiguous binding sites for FVIII separated by 5 nm: one in TIL′ and another in C8-3 that may encompass much of the face of the helical bundle containing the α1 and α3 helices (Figure 7B). Interestingly, TIL′ approaches C8-3 more closely than any other D3 module. The bipartite FVIII-binding site suggests that 2N mutations R854Q in the E′ interface with TIL′ and D879N in the Ca2+-binding loop might indirectly inhibit FVIII binding by altering TIL′–E′ and E′–D3 orientation, respectively, and disturbing the orientation between the binding sites in TIL′ and C8-3.
In conclusion, our structure of monomeric D′D3 at alkaline pH enables rational design of long half-life D′D3 complexes with FVIII for hemophilia A replacement therapy. Further structures are required to define how D′D3 dimerizes and rearranges in higher-order, helical D1D2D′D3 assemblies at acidic pH and rearranges in D′D3 dimers at alkaline pH, and how D′D3 binds FVIII.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Chao Quan and John Kulman for early contributions to this work. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the LRL Collaborative Access Team beam line facilities at Sector 31 of the Advanced Photon Source was provided by Eli Lilly & Company, which operates the facility.
Funding for this study was provided by Biogen and Bioverativ, a Sanofi company, and National Institutes of Health, National Cancer Institute grant CA-31798.
This article is dedicated to the memory of J. Evan Sadler, who contributed so much to D′D3, VWF, and our community and who died on 13 December 2018.
Authorship
Contribution: E.S.C. and J.W.A. prepared D′D3 and obtained crystals and diffraction datasets; X.D. solved the structure and built the model; X.D. and T.A.S. refined the model and made figures; X.D., N.C.L, K.E.K., Q.L., and T.A.S. analyzed data and prepared figures; X.D., N.C.L., K.E.K., R.T.P., and T.A.S. wrote initial drafts and supplemental figures; and all authors discussed the manuscript.
Conflict-of-interest disclosure: N.C.L., E.S.C., Q.L., K.E.K., and R.T.P. are employees of Bioverativ, a Sanofi company. J.W.A. is an employee of Biogen. The remaining authors declare no competing financial interests.
Correspondence: Timothy A. Springer, Center for Life Sciences, Room 3103, Boston Children’s Hospital, 3 Blackfan Cir, Boston, MA 02115; e-mail: springer_lab@crystal.harvard.edu; or Robert T. Peters, Bioverativ, 225 2nd Ave, Waltham, MA 02451; e-mail: rob.peters@bioverativ.com.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal