

The hematopoietic system is composed of over a dozen well-recognized cell types. Flow cytometry has enabled the major cell lineages to be identified and studied. However, we have increasingly appreciated that even within highly purified populations of cells, there are subtypes of cells that act on an individual basis. Studying and predicting behavior of individual cells has generally been limited by available technologies. However, enormous leaps in next-generation sequencing as well as in computational approaches have enabled single cells to be studied in unprecedented numbers. These approaches are transforming our understanding of the hematopoietic system, impacting both benign and malignant hematology. We expect these advances to substantively impact the field in the coming years, ultimately helping patients.
In this issue of Blood, we present a series of 5 reviews covering the revolutionary impact of single-cell technologies on our understanding of normal and disrupted hematopoiesis. To appreciate why single-cell approaches are so powerful, we draw an analogy of the hematopoietic system to a very precise sand art drawing, with different brightly colored sand grains representing each cell and lineage. Undisturbed, each color is distinct and easy to see, but when regions of the drawing are collected, mixed, and observed in bulk, the individual, bright colors become a drab homogeneous blend that loses the contributions of the individual grains. Single-cell technologies allow each of the individual grains (cells) to be observed on their own (Figure 1).
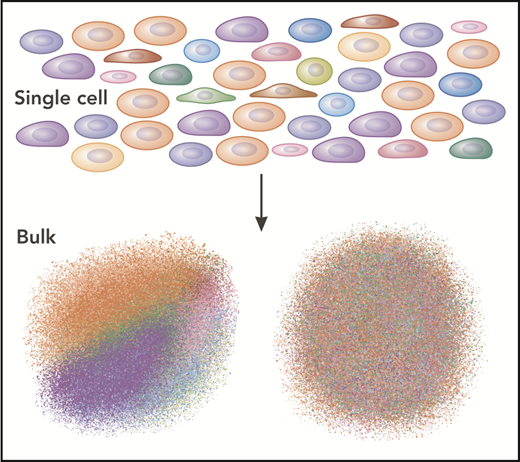
Single-cell analysis reveals the details of the population, rather than the average. The single cells are different colors here. When analyzed separately, each color (cell type) can be discerned (lower left). However, when analyzed in bulk, the “average” of all the cells dominates as a drab brown.
Single-cell analysis reveals the details of the population, rather than the average. The single cells are different colors here. When analyzed separately, each color (cell type) can be discerned (lower left). However, when analyzed in bulk, the “average” of all the cells dominates as a drab brown.
Single-cell analysis approaches are already impacting views of the relationships between cell lineages. For decades, the study of hematopoiesis has used a hierarchical model to depict the differentiation of transplantable hematopoietic stem cells into the differentiated progeny of all of the different lineages found in the peripheral blood. The hierarchical model predicts that the progeny of stem cells pass through stages where they become committed to either the lymphoid or myeloid lineages and eventually become committed to a single lineage. This model has been well supported by sophisticated in vitro and in vivo assays that have demonstrated the existence of cells capable of differentiating into colonies containing single or multiple lineages. Using these assays, cell-surface markers defining these colony-forming cells have been defined along with the epigenetic and transcriptional profiles of these populations. However, we have also known for years that hematopoiesis is more of a continuum that does not follow a strictly hierarchical progression. Single-cell approaches allow this continuum to be analyzed at a new level of detail.
In this series of reviews, the authors discuss emerging views about hematopoiesis at the single-cell level and compare these observations to the classic hierarchical model. The cumulative message from these reviews is that, although hematopoiesis generally follows a hierarchical model, the individual lineages distinguish themselves in a much more stochastic manner than we had previously imagined, especially in the context of hematologic disease.
The first review, by Loeffler and Schroeder, describes the use of single-cell imaging to track the behavior and fate of single cells. The review covers the history and contributions of time-lapse photography and its application to biology as well as the considerable challenges of single-cell tracking, particularly time frame, cell movement, and data handling. From there, the authors review more recent papers that tested the instructive vs permissive models of differentiation using single-cell tracking. Highlights include a new cell-fate determination model for GATA1 and PU.1 that may help determine whether asymmetric cell division actually occurs, a long-standing question in stem and progenitor cell biology. The review concludes with a thoughtful discussion of how these strategies can be applied to in vivo imaging and the needs for the future, which include more interdisciplinary training and more advanced statistical tools.
The other 4 reviews focus on single-cell nucleic acid profiling. This technology has been made possible by the ever-decreasing cost and ever-increasing sensitivity of high-throughput sequencing. In parallel with the increasing output of sequence data, there has been a burst of bioinformaticians who have been trained to manage and analyze the flood of data. Finally, a new culture has emerged where these data (including imaging data) are made available to the community for a greater depth of analysis that can be done in a single laboratory.
The review by Watcham, Kucinski, and Gottgens begins with an excellent description of both the methods and rationale for single-cell transcriptomics and the analytical tools, including their strengths and weaknesses. They discuss the impact of single-cell RNA sequencing (scRNAseq) on our understanding of heterogeneity within multipotent progenitor cell populations, as well as the very complicated topic of heterogeneity within the hematopoietic stem cell compartment and descriptions of the most recent paradigm-shifting data. Finally, they synthesize the state of the field into a new model and describe the future challenges for both the technology and the analytical tools.
The review by Psaila and Mead focuses more specifically on megakaryocyte and erythroid differentiation. They discuss the complications of the hierarchical model in light of recent data where transcriptional profiles of the megakaryocyte are found in bulk primitive progenitor populations. They review the data from scRNAseq studies of megakaryopoiesis and erythropoiesis and reach the consensus conclusion that megakaryocytes not only are produced in lineages shared with erythroid cells, but also can arise directly from lineage-biased stem cells. They then proceed to discuss new models derived from single-cell transcriptional profiling and offer a thoughtful look toward the future and applications to human hematopoietic disorders.
The review by Nangalia, Mitchell, and Green takes up where Psaila and Mead leave off, reviewing the role of single-cell technologies in defining the emergence of clonal hematopoiesis in hematologic disease. They begin with an excellent historical overview of tracking hematological events at a clonal level, including chromosome characterization, colony-formation assays, and murine bone marrow transplantation. They then describe the tracking of mutations in single cells and how clonal approaches have provided insights into the acquisition of somatic driver mutations, the evolution of clonal diseases, and malignant hematopoiesis.
The final review, by Gohil and Wu, focuses on how single-cell studies have transformed our understanding of chronic lymphocytic leukemia (CLL). Work described in this review is distinguished by the investigation of mutations at both the DNA and RNA levels as well as a very nice description of the role of accessory cells in CLL. Gohil and Wu have synthesized these data into an outstanding, forward-looking conclusion that uses what we have learned about the niche, subclones, and the status of the immune system to suggest clinical applications that may help guide treatment decisions.
In summary, these reviews capture a snapshot of a rapidly emerging field, encompassing both single-cell technology and applications to specific questions. We predict that the exponential growth of investigations at the single-cell level will require an altogether new set of reviews in 2 years to keep us fully abreast of how single-cell analyses have exponentially increased our understanding of normal and abnormal hematopoiesis.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal