

Key Points
Loss of the tyrosine phosphatase PTPRJ due to biallelic-null mutations in its gene causes autosomal-recessive thrombocytopenia.
Thrombocytopenia is characterized by small platelets and platelet dysfunction and derives from multiple defects in megakaryocyte biology.

Abstract
Inherited thrombocytopenias (ITs) are a heterogeneous group of disorders characterized by low platelet count that may result in bleeding tendency. Despite progress being made in defining the genetic causes of ITs, nearly 50% of patients with familial thrombocytopenia are affected with forms of unknown origin. Here, through exome sequencing of 2 siblings with autosomal-recessive thrombocytopenia, we identified biallelic loss-of-function variants in PTPRJ. This gene encodes for a receptor-like PTP, PTPRJ (or CD148), which is expressed abundantly in platelets and megakaryocytes. Consistent with the predicted effects of the variants, both probands have an almost complete loss of PTPRJ at the messenger RNA and protein levels. To investigate the pathogenic role of PTPRJ deficiency in hematopoiesis in vivo, we carried out CRISPR/Cas9-mediated ablation of ptprja (the ortholog of human PTPRJ) in zebrafish, which induced a significantly decreased number of CD41+ thrombocytes in vivo. Moreover, megakaryocytes of our patients showed impaired maturation and profound defects in SDF1-driven migration and formation of proplatelets in vitro. Silencing of PTPRJ in a human megakaryocytic cell line reproduced the functional defects observed in patients’ megakaryocytes. The disorder caused by PTPRJ mutations presented as a nonsyndromic thrombocytopenia characterized by spontaneous bleeding, small-sized platelets, and impaired platelet responses to the GPVI agonists collagen and convulxin. These platelet functional defects could be attributed to reduced activation of Src family kinases. Taken together, our data identify a new form of IT and highlight a hitherto unknown fundamental role for PTPRJ in platelet biogenesis.
Introduction
Inherited thrombocytopenias (ITs) are a heterogeneous group of disorders characterized by low platelet count that may result in bleeding tendency. Some ITs are characterized only by a platelet defect, consisting of thrombocytopenia, with or without alterations in platelet function (nonsyndromic ITs). In other forms, thrombocytopenia associates with additional congenital defects that affect different tissues and organs (syndromic ITs). Finally, some disorders present with isolated thrombocytopenia but are characterized by the propensity to develop additional manifestations over time (predisposing ITs).1 Another classification of ITs is based on platelet size, which is useful in clinical practice for differential diagnosis among the different forms; in some cases, it may provide clues for distinguishing genetic from acquired thrombocytopenia.2-4
Despite recent progress in the field, ITs remain poorly understood. Nearly 50% of patients with familial thrombocytopenia are affected with forms of unknown origin, because they do not fit the criteria for any known disorder and do not carry mutations in known IT genes.5,6 The identification of the molecular basis of thrombocytopenia in these cases is an essential prerequisite for providing patients with a definite diagnosis and tailored clinical management, including access to studies that test emerging therapeutic approaches. Moreover, the discovery of new IT genes provides novel information about the mechanisms of megakaryopoiesis and platelet biogenesis.7,8
We provide evidence that biallelic loss-of-function mutations in the PTPRJ gene cause a new form of IT. The disorder is associated with the almost complete loss of PTPRJ expression and presents as a nonsyndromic IT that is characterized by small platelets and platelet functional defects. Investigation of patients’ megakaryocytes suggested that thrombocytopenia derives from multiple alterations in megakaryocyte function.
Patients and methods
Patients
Clinical investigation of the family members was performed at the Pediatric Unit, University “Aldo Moro” and the Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Policlinico San Matteo Foundation. The study was approved by the Institutional Review Board of Pavia. All investigated individuals or their legal guardians provided written informed consent for the study, which was conducted in accordance with the Declaration of Helsinki.
Whole-exome sequencing and variants follow-up
Whole-exome sequencing on genomic DNA samples and raw data analysis were performed as described.5 Candidate variants were selected and confirmed through Sanger sequencing following the criteria and procedures detailed in supplemental Methods (available on the Blood Web site).
Platelet studies
Platelet size was measured by software-assisted image analysis on blood smears, as described.2 Protocol and reagents used in studies of platelet aggregation and activation in response to agonists,9,10 as well as platelet flow cytometry,9,10 are reported in supplemental Methods. A platelet-aggregation assay and studies requiring washed platelets were not possible in proband I-1 because of the very low platelet count.
Immunoblotting
Preparation of washed platelet lysates or cell lysates and immunoblotting procedures were described previously.11,12 Antibodies are listed in supplemental Methods. Densitometric analysis of bands was performed using ImageJ software (https://imagej.nih.gov/ij/). Results represent the mean ± standard deviation (SD) of 3 separate immunoblotting experiments.
PTPRJ RNA analysis
RNA was extracted from peripheral blood or transduced Dami cells, and PTPRJ expression was evaluated using real-time polymerase chain reaction (PCR) and expressed as fold-change expression relative to ACTB, as detailed in supplemental Methods. Results represent the mean ± standard deviation (SD) of 3 independent experiments.
Megakaryocyte studies
Megakaryocytes were differentiated from peripheral blood progenitors by 14-day culture as described.13,14 Megakaryocyte differentiation and maturation at the end of the culture were investigated by flow cytometry as reported.14 Proplatelet formation was studied.15,16 SDF1-driven migration was investigated using a Transwell migration chamber system (Merck Millipore, Milan, Italy).15,17 Protocols are detailed in supplemental Methods. Samples from the patients were processed and analyzed simultaneously with those of 3 age-matched healthy individuals.
Zebrafish and embryo maintenance
All embryos were collected from natural matings of adult zebrafish (Danio rerio) on the cd41:GFP background18 and maintained in egg water at 28.5°C until 4 days postfertilization (dpf).
CRISPR gRNA generation and efficiency
Ensembl (http://www.ensembl.org) was exploited to identify the zebrafish PTPRJ ortholog, ptprja (ENSDARG00000033042). CRISPR guide RNA (gRNA) targets and flanking primers were designed using ChopChop19,20 (http://chopchop.cbu.uib.no/) and synthesized as detailed in supplementary methods. CRISPR/Cas9 targeting efficiency was calculated as the total number of clones per embryo with small insertions or deletions, and overall gRNA efficiency was calculated as the average of all embryo efficiencies associated with that gRNA.
Thrombocyte imaging
Embryos were injected at the 1-cell stage within 1 hour postfertilization with CRISPR gRNA, with or without Cas9 protein. Embryos were imaged on an agarose gel mold at 4 dpf for thrombocyte counting. Cells were quantified in the ventral portion of the tail from the end of the yolk to the tip of the tail, according to the protocols detailed in supplemental Methods.
Cell line
Dami cells were kindly provided by Stefania Rigacci (University of Florence, Florence, Italy). The phenotype of these cells was characterized extensively and reevaluated periodically.12
Lentiviral constructs and cell transduction
Lentiviral particles were obtained using the pLKO.1 TRC vector kindly provided by The RNAi Consortium.21 Protocols for preparation of lentiviral suspensions, Dami cell transduction, and selection are described in supplemental Methods. PTPRJ knockdown was routinely checked by real-time PCR and immunoblotting.
Functional studies on cell lines
Results
Clinical presentation of the family
Main clinical and hematological features of the investigated family are summarized in Table 1. The probands were a 15-year-old girl (II-1) and her 9-year-old brother (II-2) who were referred for investigation of congenital thrombocytopenia (Figure 1A). Their parents had normal platelet counts, and no other siblings are present. Proband II-1 had a history of spontaneous bleeding consisting of menorrhagia, easy bruising, petechiae, and epistaxis, resulting in mild iron-deficiency anemia. Individual II-2 also presented with spontaneous bleeding, although of a milder degree. With the exception of bleeding tendency, the medical history of both probands was unremarkable, and physical examination did not reveal any relevant abnormalities.
Main clinical and hematological features of the investigated family
| Subject | PTPRJ genotype | Gender/age, y | Platelets, ×109/L | MPV, fL* | Hgb, g/dL | MCV, fL | WBC, ×109/L | Neu, ×109/L | ISTH BAT score† | Bleeding episodes | Other findings |
|---|---|---|---|---|---|---|---|---|---|---|---|
| II-1 | c.97-2A>G/c.1875delG | F/15 | 12 | 6.7 | 10.6 | 69.9 | 6.4 | 4.4 | 9 | Easy bruising, petechiae, menorrhagia, epistaxis | Iron deficiency, valgus knee |
| II-2 | c.97-2A>G/c.1875delG | M/9 | 83 | 6.8 | 12.6 | 81.0 | 6.4 | 3.2 | 5 | Easy bruising, epistaxis | None |
| I-1 | c.97-2A>G/WT | M/48 | 250 | 7.5 | 14.2 | 89.6 | 5.1 | 3.1 | 0 | None | None |
| I-2 | c.1875delG/WT | F/44 | 201 | 7.8 | 12.0 | 89.7 | 4.5 | 2.8 | 0 | None | None |
| Subject | PTPRJ genotype | Gender/age, y | Platelets, ×109/L | MPV, fL* | Hgb, g/dL | MCV, fL | WBC, ×109/L | Neu, ×109/L | ISTH BAT score† | Bleeding episodes | Other findings |
|---|---|---|---|---|---|---|---|---|---|---|---|
| II-1 | c.97-2A>G/c.1875delG | F/15 | 12 | 6.7 | 10.6 | 69.9 | 6.4 | 4.4 | 9 | Easy bruising, petechiae, menorrhagia, epistaxis | Iron deficiency, valgus knee |
| II-2 | c.97-2A>G/c.1875delG | M/9 | 83 | 6.8 | 12.6 | 81.0 | 6.4 | 3.2 | 5 | Easy bruising, epistaxis | None |
| I-1 | c.97-2A>G/WT | M/48 | 250 | 7.5 | 14.2 | 89.6 | 5.1 | 3.1 | 0 | None | None |
| I-2 | c.1875delG/WT | F/44 | 201 | 7.8 | 12.0 | 89.7 | 4.5 | 2.8 | 0 | None | None |
F, female; Hgb, hemoglobin; M, male; Neu, neutrophils; WBC, white blood cells; WT, wild type.
Evaluated using an automated cell counter, normal range 7-13 fL.
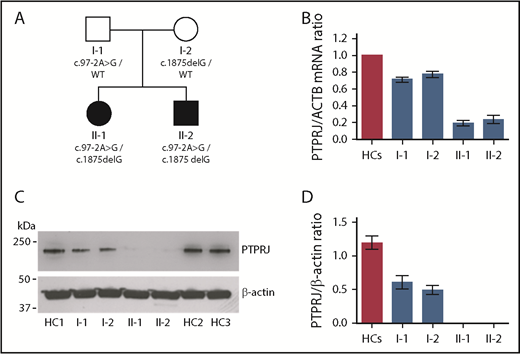
Compound heterozygosity for the variants identified in PTPRJ results in the almost complete loss of mRNA and protein. (A) Pedigree of the investigated family. Black symbols indicate thrombocytopenia. (B) PTPRJ mRNA expression in whole blood of the members of the family, as detected by real-time PCR. Expression is reported as fold change in PTPRJ relative to ACTB (β-actin) levels calculated with the ΔΔCt method. Data represent the means ± SD of 3 independent experiments using 3 HCs. (C-D) PTPRJ protein abundance in platelets from the members of the family. Lysates of washed platelets obtained from the 2 probands, their parents, and different HCs were analyzed by immunoblotting using an antibody against PTPRJ. β-actin was used as loading control. (C) Representative image of the immunoblotting experiments. (D) Densitometric analysis of the bands obtained in 3 independent experiments (means ± SD). PTPRJ level is expressed as the PTPRJ/β-actin ratio.
Compound heterozygosity for the variants identified in PTPRJ results in the almost complete loss of mRNA and protein. (A) Pedigree of the investigated family. Black symbols indicate thrombocytopenia. (B) PTPRJ mRNA expression in whole blood of the members of the family, as detected by real-time PCR. Expression is reported as fold change in PTPRJ relative to ACTB (β-actin) levels calculated with the ΔΔCt method. Data represent the means ± SD of 3 independent experiments using 3 HCs. (C-D) PTPRJ protein abundance in platelets from the members of the family. Lysates of washed platelets obtained from the 2 probands, their parents, and different HCs were analyzed by immunoblotting using an antibody against PTPRJ. β-actin was used as loading control. (C) Representative image of the immunoblotting experiments. (D) Densitometric analysis of the bands obtained in 3 independent experiments (means ± SD). PTPRJ level is expressed as the PTPRJ/β-actin ratio.
Identification and characterization of the PTPRJ variants
The inheritance pattern of thrombocytopenia in this family was consistent with a recessive disorder. By performing whole-exome sequencing of probands II-1 and II-2, we identified 6 genes with 2 heterozygous variants each; no homozygous alleles were observed. Segregation analysis showed that only the 2 variants in PTPRJ were in trans and consistent with an autosomal-recessive inheritance (Figure 1A; supplemental Table 1). The g.48131608A>G (c.97-2A>G) affects the splice acceptor site of intron 1 and is predicted to cause skipping of exon 2 in the messenger RNA (mRNA), whereas the g.48158556delG (c.1875delG) affects the first nucleotide of exon 10. By complementary DNA sequencing, we confirmed that both variants result in frameshift and insertion of a premature stop codon, causing r.97_115del19 (p.Thr38Profs9X) and r.1875delG (p.Ser626Alafs7X), respectively. A variant in position g.48131608, but with a different nucleotide change (A>C), has been reported once (in heterozygosity) in the gnomAD dataset (rs758226104, MAF 4.063e-6) (http://gnomad.broadinstitute.org/). No homozygous-null variants in PTPRJ are described in The Genome Aggregation Database or the Exome Aggregation Consortium (http://exac.broadinstitute.org).
Direct testing of the effect of the mutations was consistent with predictions. Real-time PCR analysis of RNA from whole blood showed that PTPRJ mRNA is severely depleted in the 2 probands and moderately downregulated in their parents, suggesting that the mutations cause mRNA decay (Figure 1B). Immunoblotting of platelets from the probands showed no appreciable detection of PTPRJ protein, whereas platelets from the parents presented an ∼50% expression compared with healthy controls (HCs) (Figure 1C-D).
Platelet phenotype
Examination of blood smears after conventional staining demonstrated a striking presence of small-sized platelets in probands II-1 and II-2 (Figure 2A). Image analysis of platelet size using an established method2 confirmed that the probands present a reduced mean platelet diameter and an increased proportion of small platelets (platelets smaller than the 2.5th percentile of platelet diameter distribution in healthy subjects) (supplemental Table 2). Platelet size was normal in both parents. No other relevant morphological abnormalities in platelets or other blood cells were found in the 4 family members using light microscopy or transmission electron microscopy analysis (supplemental Figure 1).
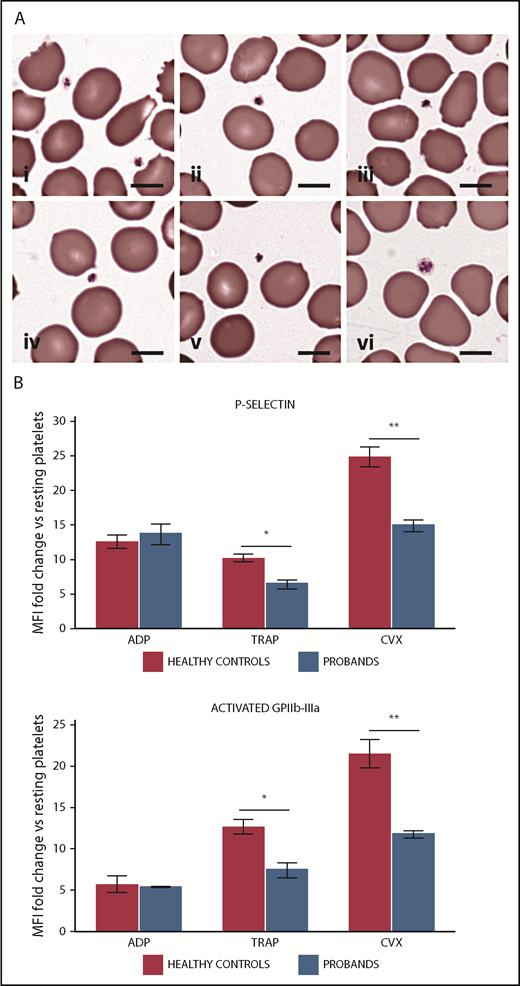
Patients with PTPRJ-null variants present platelets with small size and defective platelet response to convulxin and thrombin receptor activating peptide (TRAP). (A) Peripheral blood smears, May-Grünwald-Giemsa staining: representative examples of small-sized platelets from proband I-2 (i-iii) and proband II-2 (iv-v). The images are representative of the average platelet observable in patients’ blood smears. In (vi), an average platelet of normal size for a healthy subject is shown for comparison. Scale bars, 5 µm. (B) Flow cytometry of platelet activation in response to ADP, TRAP, and convulxin (CVX) in probands II-1 and II-2. Platelet surface expression of P-selectin and of the activated form of GPIIb-IIIa (PAC1 antibody binding) was measured after incubation with ADP (5 mM), TRAP (25 µM), CVX (100 ng/mL), or vehicle (HEPES buffer). Platelet activation is expressed as the ratio between mean fluoresce intensity (MFI) measured after stimulation with each agonist and MFI measured after incubation with the buffer alone (resting platelets). The values obtained in the 2 probands were aggregated and compared with those of 3 HCs processed in parallel. Data represent the mean ± SD of 2 independent analyses. *P < .05, **P < .01, 2-tailed Student t test.
Patients with PTPRJ-null variants present platelets with small size and defective platelet response to convulxin and thrombin receptor activating peptide (TRAP). (A) Peripheral blood smears, May-Grünwald-Giemsa staining: representative examples of small-sized platelets from proband I-2 (i-iii) and proband II-2 (iv-v). The images are representative of the average platelet observable in patients’ blood smears. In (vi), an average platelet of normal size for a healthy subject is shown for comparison. Scale bars, 5 µm. (B) Flow cytometry of platelet activation in response to ADP, TRAP, and convulxin (CVX) in probands II-1 and II-2. Platelet surface expression of P-selectin and of the activated form of GPIIb-IIIa (PAC1 antibody binding) was measured after incubation with ADP (5 mM), TRAP (25 µM), CVX (100 ng/mL), or vehicle (HEPES buffer). Platelet activation is expressed as the ratio between mean fluoresce intensity (MFI) measured after stimulation with each agonist and MFI measured after incubation with the buffer alone (resting platelets). The values obtained in the 2 probands were aggregated and compared with those of 3 HCs processed in parallel. Data represent the mean ± SD of 2 independent analyses. *P < .05, **P < .01, 2-tailed Student t test.
Flow cytometry did not show any consistent defect in the expression of the major glycoproteins of the platelet surface in any family member (supplemental Table 3).
Analysis of platelet aggregation in proband II-2 showed defective response to collagen (4 µg/mL) and to the GPVI-specific agonist convulxin (60 ng/mL). The defect was overcome at higher concentrations of these agonists (Table 2). The patient also exhibited a mildly reduced platelet aggregation after stimulation with TRAP (25 µM), whereas aggregation was normal in response to adenosine 5′-diphosphate (ADP) (5 µM) and ristocetin (1.5 mg/mL). Platelet functional response to ADP, convulxin, and TRAP was also assessed as the induction of surface expression of P-selectin and the activated form of GPIIb-IIIa.23 Consistent with the findings of the aggregation assay, platelets from probands II-1 and II-2 showed a reduced P-selectin exposure and GPIIb-IIIa activation after stimulation with convulxin and TRAP, whereas response to ADP was normal (Figure 2B). The probands’ parents exhibited normal platelet responses to all of the tested agonists (Table 2, and data not shown).
In vitro platelet aggregation in the investigated subjects, maximal extent (percentage)
| Subject | Collagen, 4 µg/mL | Collagen, 20 µg/mL | Convulxin, 60 ng/mL | Convulxin, 100 ng/mL | ADP, 5 μM | TRAP, 25 μM | Ristocetin, 1.5 mg/mL |
|---|---|---|---|---|---|---|---|
| II-2 | 43 | 89 | 43 | 77 | 81 | 61 | 90 |
| I-1 | 97 | nd | 89 | nd | 86 | 100 | 100 |
| I-2 | 70 | nd | 97 | nd | 90 | 100 | 96 |
| Subject | Collagen, 4 µg/mL | Collagen, 20 µg/mL | Convulxin, 60 ng/mL | Convulxin, 100 ng/mL | ADP, 5 μM | TRAP, 25 μM | Ristocetin, 1.5 mg/mL |
|---|---|---|---|---|---|---|---|
| II-2 | 43 | 89 | 43 | 77 | 81 | 61 | 90 |
| I-1 | 97 | nd | 89 | nd | 86 | 100 | 100 |
| I-2 | 70 | nd | 97 | nd | 90 | 100 | 96 |
Normal ranges: collagen, 66-88%; convulxin, 70-100%; ADP, 43-76%; TRAP, 70-100%; ristocetin, 67-90%.
nd, not determined.
We were able to characterize the GPVI-mediated functional defect further by analyzing tyrosine phosphorylation after stimulation with convulxin in platelets from proband II-2. We observed a marked inhibition of the increase in tyrosine phosphorylation of all detectable proteins in response to convulxin over a 300-second time course (Figure 3A-B). In Ptprj-knockdown mice, Senis and colleagues found a similar impaired response to GPVI stimulation, which could be partially explained by an ∼60% reduction in GPVI expression.24,25 In our patients, GPVI expression was normal (supplemental Figure 2; supplemental Table 3), suggesting that the functional defect is entirely attributable to impaired signaling downstream of the receptor. Investigation of in vitro models and Ptprj-ablated mice suggested that the defective GPVI signaling is caused by reduced activation of Src family kinases (SFKs) as a consequence of the loss of PTPRJ. By dephosphorylating the inhibitory tyrosine residues of SFKs, Ptprj maintains a baseline level of activation of SFKs in resting platelets, which is required for a physiologic response to stimulation of the collagen receptors.24-26 Consistent with this model, we found that the SFK SRC was in a state of markedly reduced activation in platelets from patient II-2 (increased phosphorylation of the inhibitory tyrosine 530 and parallel reduced phosphorylation of the activation tyrosine 419) in resting conditions and after stimulation with convulxin (Figure 3A,C). Consistent with the reduced activation of SFKs, the activation of the tyrosine kinase Syk in response to convulxin was markedly reduced (supplemental Figure 3).
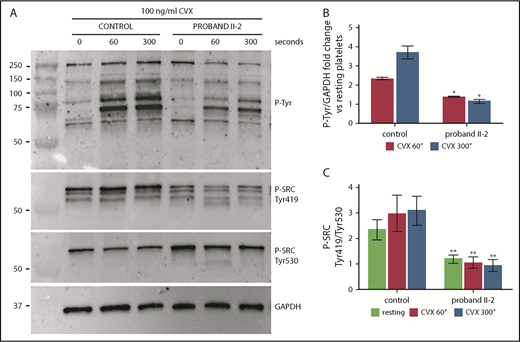
Platelets from proband II-2 show impaired tyrosine phosphorylation after stimulation with convulxin (CVX) and reduced activation of the SFK SRC. Platelets were obtained from peripheral blood from proband II-2 and HCs. Lysates were prepared with resting platelets (time 0 seconds) and after stimulation with 100 ng/mL CVX for 60 and 300 seconds. Immunoblotting was performed with the 4G10 anti-phosphotyrosine antibody (P-Tyr), with an antibody recognizing SRC phosphorylated at Tyr419 (SRC activation tyrosine) and with an antibody recognizing SRC phosphorylated at Tyr530 (SRC inhibitory tyrosine). GAPDH was used as loading control. (A) Representative image of the immunoblotting experiments. (B) Densitometric analysis of tyrosine phosphorylation. The bands obtained with 3 separate experiments (mean ± SD) were analyzed. Change in tyrosine phosphorylation after stimulation with CVX for 60 and 300 seconds is expressed as the fold increase in the P-tyrosine/GAPDH ratio with respect to the resting condition. The proband showed a marked inhibition of the increase in tyrosine phosphorylation of all detectable proteins in response to CVX at both time points. (C) Densitometric analysis of SRC phosphorylation. The bands obtained in 3 separate experiments (means ± SD) were analyzed. SRC activation status is expressed as the SRC phospho-Tyr419/SRC phospho-Tyr530 ratio normalized to GAPDH. The proband showed significantly reduced activation of SRC under resting conditions and after activation with CVX. *P < .05, **P < .01 vs HCs, 2-tailed Student t test.
Platelets from proband II-2 show impaired tyrosine phosphorylation after stimulation with convulxin (CVX) and reduced activation of the SFK SRC. Platelets were obtained from peripheral blood from proband II-2 and HCs. Lysates were prepared with resting platelets (time 0 seconds) and after stimulation with 100 ng/mL CVX for 60 and 300 seconds. Immunoblotting was performed with the 4G10 anti-phosphotyrosine antibody (P-Tyr), with an antibody recognizing SRC phosphorylated at Tyr419 (SRC activation tyrosine) and with an antibody recognizing SRC phosphorylated at Tyr530 (SRC inhibitory tyrosine). GAPDH was used as loading control. (A) Representative image of the immunoblotting experiments. (B) Densitometric analysis of tyrosine phosphorylation. The bands obtained with 3 separate experiments (mean ± SD) were analyzed. Change in tyrosine phosphorylation after stimulation with CVX for 60 and 300 seconds is expressed as the fold increase in the P-tyrosine/GAPDH ratio with respect to the resting condition. The proband showed a marked inhibition of the increase in tyrosine phosphorylation of all detectable proteins in response to CVX at both time points. (C) Densitometric analysis of SRC phosphorylation. The bands obtained in 3 separate experiments (means ± SD) were analyzed. SRC activation status is expressed as the SRC phospho-Tyr419/SRC phospho-Tyr530 ratio normalized to GAPDH. The proband showed significantly reduced activation of SRC under resting conditions and after activation with CVX. *P < .05, **P < .01 vs HCs, 2-tailed Student t test.
Finally, platelets from the 2 probands presented normal WASP and c-MPL expression (supplemental Figure 4), suggesting that PTPRJ loss does not affect these pathways, whose defects are associated with the finding of small platelets in other forms of IT.2
Megakaryocyte phenotype
To investigate the mechanisms of thrombocytopenia, we differentiated megakaryocytes in vitro from peripheral blood progenitors from the 2 probands according to a standard protocol.13,14 Megakaryocyte differentiation, evaluated as the yield of CD41+ cells at the end of the culture,13 was not different between the patients and 3 healthy subjects processed in parallel (Figure 4A-B). However, the proportion of mature megakaryocytes, measured as the percentage of CD41+ cells coexpressing CD42b,14 was slightly, but significantly, lower in patients (Figure 4C), suggesting a mild defect in terminal megakaryocyte maturation.
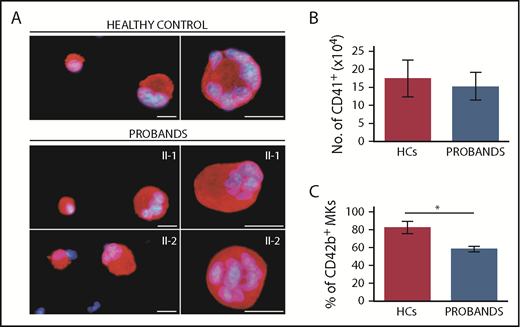
Megakaryocytes (MKs) of the probands exhibit normal in vitro differentiation and defective terminal maturation. MKs were differentiated from peripheral blood progenitor cells through 14-day culture. Samples from probands II-1 and II-2 were processed in parallel with those of 3 HCs. (A) Representative images of MKs labeled with an anti–β1-tubulin antibody (red fluorescence). Hoechst (blue) was used for counterstaining nuclei. Scale bars, 25 µm. (B) MK differentiation was assessed as the yield (absolute number) of CD41+ cells at day 14 of culture, as measured by flow cytometry. (C) MK maturation was assessed as the percentage of CD41+ cells coexpressing the CD42b antigen, as measured by flow cytometry. *P < .001, 2-tailed Student t test.
Megakaryocytes (MKs) of the probands exhibit normal in vitro differentiation and defective terminal maturation. MKs were differentiated from peripheral blood progenitor cells through 14-day culture. Samples from probands II-1 and II-2 were processed in parallel with those of 3 HCs. (A) Representative images of MKs labeled with an anti–β1-tubulin antibody (red fluorescence). Hoechst (blue) was used for counterstaining nuclei. Scale bars, 25 µm. (B) MK differentiation was assessed as the yield (absolute number) of CD41+ cells at day 14 of culture, as measured by flow cytometry. (C) MK maturation was assessed as the percentage of CD41+ cells coexpressing the CD42b antigen, as measured by flow cytometry. *P < .001, 2-tailed Student t test.
Migration of megakaryocytes in the bone marrow (BM) to areas enriched in vessels is essential for efficient release of platelets into the circulation; SDF1 is the key chemotactic agent driving this process.27-29 Therefore, we investigated the ability of patients’ megakaryocytes to migrate toward an SDF1 gradient using a Transwell assay.15,17 Megakaryocytes from the probands exhibited a profound defect in migration on type I collagen and fibrinogen, 2 of the most abundant proteins of the BM extracellular matrix (ECM) (Figure 5A-C).30,31
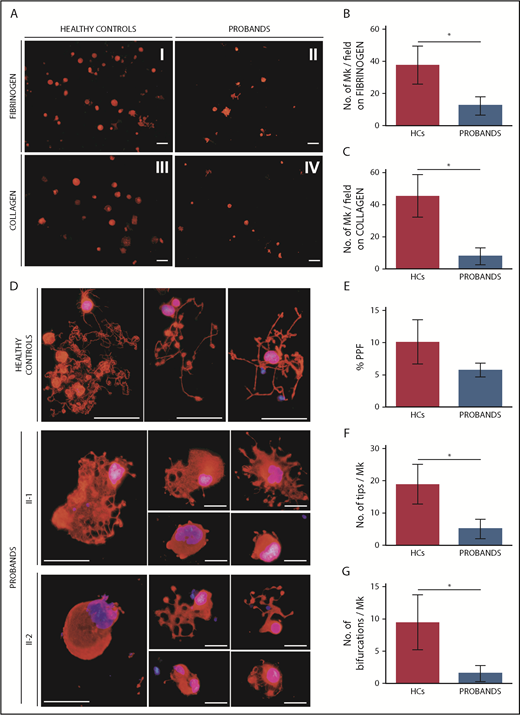
Megakaryocytes (MKs) of the probands exhibit defective SDF1-driven migration on fibrinogen and type I collagen and altered proplatelet formation. (A-C) SDF1-driven migration of MKs was investigated using a Transwell assay. Transwell systems having a polycarbonate membrane with an 8-µm pore size were coated with fibrinogen or type I collagen. Aliquots of 1 × 104 MKs were seeded in the upper chamber of the Transwell insert, whereas the lower chamber was filled with medium containing 100 ng/mL SDF1. After incubation for 16 hours at 37°C and 5% CO2, cells that migrated to the lower face of the membrane were labeled with an anti–β1-tubulin antibody (red) and counted using fluorescence microscopy with an Olympus BX-51 microscope. Samples of the 2 probands were processed in parallel with those of 3 HCs. (A) Representative images of microscopic fields of migrated cells. Scale bars, 30 µm. (B-C) MK migration was quantified as the number of migrated MKs per field (mean ± SD) by analyzing the entire polycarbonate membrane area. The assays were performed in triplicate wells for each condition. (D-G) Proplatelet formation was analyzed on fibrinogen-coated coverslips after incubation of MKs for 16 hours at 37°C and 5% CO2. Cells were stained with an anti–β1-tubulin antibody (red). Hoechst (blue) was used for counterstaining nuclei. Samples of the probands were processed in parallel with those of 3 HCs. (D) Representative images of the morphology of proplatelets extended by probands II-1 and II-2. Proplatelets from HCs are shown in the top row comparison. Scale bars, 30 µm. (E) The rate of proplatelet formation (%PPF) was measured, using fluorescence microscopy, as the proportion of MKs displaying ≥1 proplatelet with respect to the total number of MKs (mean ± SD). The number of proplatelet free ends (tips) (F) and the number of bifurcations of proplatelet shafts (G) per MK were measured using image analysis that investigated ≥25 MKs for each individual (patient or control). *P < .0001, 2-tailed Student t test.
Megakaryocytes (MKs) of the probands exhibit defective SDF1-driven migration on fibrinogen and type I collagen and altered proplatelet formation. (A-C) SDF1-driven migration of MKs was investigated using a Transwell assay. Transwell systems having a polycarbonate membrane with an 8-µm pore size were coated with fibrinogen or type I collagen. Aliquots of 1 × 104 MKs were seeded in the upper chamber of the Transwell insert, whereas the lower chamber was filled with medium containing 100 ng/mL SDF1. After incubation for 16 hours at 37°C and 5% CO2, cells that migrated to the lower face of the membrane were labeled with an anti–β1-tubulin antibody (red) and counted using fluorescence microscopy with an Olympus BX-51 microscope. Samples of the 2 probands were processed in parallel with those of 3 HCs. (A) Representative images of microscopic fields of migrated cells. Scale bars, 30 µm. (B-C) MK migration was quantified as the number of migrated MKs per field (mean ± SD) by analyzing the entire polycarbonate membrane area. The assays were performed in triplicate wells for each condition. (D-G) Proplatelet formation was analyzed on fibrinogen-coated coverslips after incubation of MKs for 16 hours at 37°C and 5% CO2. Cells were stained with an anti–β1-tubulin antibody (red). Hoechst (blue) was used for counterstaining nuclei. Samples of the probands were processed in parallel with those of 3 HCs. (D) Representative images of the morphology of proplatelets extended by probands II-1 and II-2. Proplatelets from HCs are shown in the top row comparison. Scale bars, 30 µm. (E) The rate of proplatelet formation (%PPF) was measured, using fluorescence microscopy, as the proportion of MKs displaying ≥1 proplatelet with respect to the total number of MKs (mean ± SD). The number of proplatelet free ends (tips) (F) and the number of bifurcations of proplatelet shafts (G) per MK were measured using image analysis that investigated ≥25 MKs for each individual (patient or control). *P < .0001, 2-tailed Student t test.
Finally, we studied the ability of megakaryocytes to form proplatelets. The patients had a reduced proportion of megakaryocytes extending proplatelets in adhesion to fibrinogen, a substrate that promotes proplatelet formation (Figure 5D-E).13,14 Moreover, patients’ proplatelets exhibited an altered morphology that was characterized by reduced length and ramification of their shafts, resulting in a smaller number of proplatelet free ends (Figure 5D,F-G). Of note, the same alterations in proplatelet formation were also evident when megakaryocytes were cultured in suspension (supplemental Figure 5), suggesting that these defects are intrinsic to megakaryocytes and are independent of the interaction with specific ECM proteins.
ptprj ablation in zebrafish
To assess whether the absence of PTPRJ induces thrombocytopenia in vivo, we took advantage of zebrafish as an established model for testing gene and allele function.32 Previous studies have shown that loss-of-function mutations in bona fide thrombocytopenia-causing genes lead to a reduction in CD41+ cells.18,33,34 We used CRISPR/Cas9 to ablate ptprj in a transgenic line expressing GFP under the control of a CD41 promoter (cd41:GFP18 ) to determine whether this manipulation might affect the production of CD41+ cells.
Reciprocal BLAST searches with human PTPRJ identified 3 possible zebrafish orthologs (ptprja, ptprjb.1, and ptprjb.2). Further inspection of each predicted transcript failed to identify a start methionine in ptprjb.1 or ptprjb.2; moreover, both “b” transcripts did not encode the fibronectin repeats of the human ortholog (supplemental Figure 6A-B). Together, these observations suggested that ptprja is the likely sole functional ortholog of human PTPRJ. As such, we focused on this transcript for our downstream studies. To target this locus, we identified a gRNA target site on exon 4 (G4), selected based on estimated efficiency, guanine-cytosine content, and complementarity. Efficiency testing by heteroduplex analysis35 showed that injected embryos had a high level of mosaicism (supplemental Figure 6C); this was substantiated by direct sequencing of the locus, which showed that 83% of transcripts produced by the locus contained nested deletions, almost all of which induced frame shifts (supplemental Figure 6D).
To test whether F0 mutants display a depletion of CD41+ cells, we imaged and counted GFP+ cells in a consistent region at 4 dpf, defined by unambiguous anatomical landmarks (the end of the yolk to the tip of the tail; Figure 6). Injection of 50 pg of guide mRNA with CAS9 protein induced a significant and reproducible depletion of CD41+ cells compared with wild-type age-matched embryos or with embryos injected with gRNA and no enzyme (P < 5.1 × 10−7; Figure 6). Repetition of the experiment at a higher dose (100 pg of gRNA+CAS9) replicated the phenotype (P < 8 × 10−5; supplemental Figure 7). To confirm that the thrombocytopenia was a result of ptprja editing and not off-target effects, we designed a second guide RNA (G1) with a nonoverlapping PAM site that targeted the same exon with high mosaicism (73% of transcripts edited; supplemental Figure 6C). Injection of the second guide RNA with CAS9 recapitulated the thrombocytopenia phenotype, thereby supporting the specificity of ptprja CRISPR targeting (P < 4 × 10−5; supplemental Figure 7). Together, these results corroborate the human genetic and expression data, suggesting that PTPRJ is necessary for thrombocyte development.
![Figure 6. Ablation of PTPRJ zebrafish ortholog, ptprja, results in thrombocyte reductions. (A) Representative images of larvae at 4 dpf for each condition expressing transgenic cd41:GFP. Scale bar, 100 µm. (B) Number of thrombocytes counted at a consistent location on lateral images of the body [red rectangle in (A)], for each condition: uninjected controls and 50 pg G4, with or without 200 pg Cas9. ****P < .0001, 2-tailed Student t test.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/12/10.1182_blood-2018-07-859496/4/m_blood859496f6.png?Expires=1769079444&Signature=ePfbRRl9GWf4foHYSUnZRYy1boAf~hkaAzUOck5-SlGIr3lsB05Pe4XTPmtOh-1l0QFBAE2YyQvTrUu5zQ9d5zxCSFdnpNj~GmPvI4hhmd~OoDwjCWs33Mn-tBzlmG809TDznGaO-MW7tqmMw6fQMW29KCOIoITtnqN9GqsEKcGpNERu6sg8hGgXdtUywBecjqnUcnBRHvDeCmACC2aKNPN~sy9uchVpmLvsDMDZRmTQXlva~86VrsmwOEEU2BEpSPD~H0Wpavr~VC0zxWumdsubA0ZKN2AsFA5RTs7gtFvxEb8RkFYkAV1H1sLX3oP2rU5MO8eWBgRcLfmph8012Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Ablation of PTPRJ zebrafish ortholog, ptprja, results in thrombocyte reductions. (A) Representative images of larvae at 4 dpf for each condition expressing transgenic cd41:GFP. Scale bar, 100 µm. (B) Number of thrombocytes counted at a consistent location on lateral images of the body [red rectangle in (A)], for each condition: uninjected controls and 50 pg G4, with or without 200 pg Cas9. ****P < .0001, 2-tailed Student t test.
Ablation of PTPRJ zebrafish ortholog, ptprja, results in thrombocyte reductions. (A) Representative images of larvae at 4 dpf for each condition expressing transgenic cd41:GFP. Scale bar, 100 µm. (B) Number of thrombocytes counted at a consistent location on lateral images of the body [red rectangle in (A)], for each condition: uninjected controls and 50 pg G4, with or without 200 pg Cas9. ****P < .0001, 2-tailed Student t test.
PTPRJ ablation in a human megakaryocytic cell line
We also investigated the effects of PTPRJ knockdown in Dami cells, a well-characterized human megakaryocytic cell line12,36,37 that expresses PTPRJ constitutively. By lentiviral transduction of a pool of short hairpin RNAs (shRNAs), we generated Dami cells stably expressing <10% of the PTPRJ protein compared with cells transduced with the control vector (supplemental Figure 8). A Transwell assay demonstrated that PTPRJ knockdown induces a significant inhibition of SDF1-driven migration of Dami cells on type I collagen and fibrinogen, reproducing the defect observed in patients’ megakaryocytes (Figure 7A-B). Incubation of Dami cells with thrombopoietin and phorbol 12-myristate 13-acetate promotes maturation of these cells toward the megakaryocytic lineage, which can be quantified by the increase in surface expression of megakaryocyte-specific markers.22 After the treatment, PTPRJ-knockdown Dami cells exhibited a similar increase in surface CD61 and CD41 expression compared with control cells but a defective increase in the late megakaryocyte maturation marker CD42b (Figure 7C). The proportion of CD41+ cells coexpressing CD42b after thrombopoietin/phorbol 12-myristate 13-acetate treatment was lower in PTPRJ-knockdown cells, suggesting that PTPRJ knockdown induces a defect in the late megakaryocyte maturation, similar to what was observed in patients’ megakaryocytes.
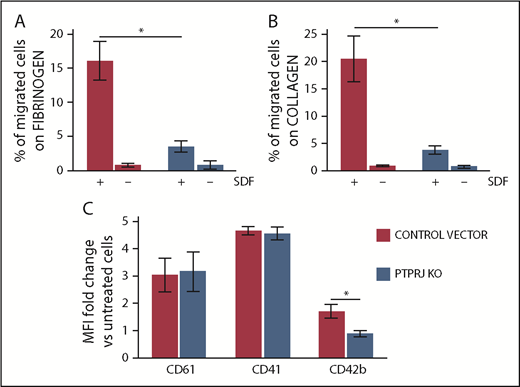
PTPRJ knockdown results in inhibition of SDF1-driven migration of the megakaryocytic Dami cell line, as well as in reduced thrombopoietin and phorbol-induced maturation of Dami cells. Using lentiviral transduction of a pool of shRNA, we generated Dami cells expressing <10% PTPRJ protein (PTPRJ KO) compared with cells transduced with the negative control vector. (A-B) SDF1-driven migration of Dami cells was investigated using a Transwell assay. Transwell systems having a polycarbonate membrane with an 8-µm pore size were coated with fibrinogen (A) or type I collagen (B). Aliquots of 1 × 105 cells were seeded in the upper chamber of the Transwell system, whereas medium with 300 ng/mL SDF1 (+) or without SDF1 (–) (negative control conditions) was added to the lower chamber. After incubation for 16 hours at 37°C and 5% CO2, the percentage of cells that migrated to the lower chamber with respect to the number of cells put in the upper chamber at the beginning of the experiment was calculated. Each experiment was performed in duplicate, and data represent the mean of 3 separate experiments. *P < .01, 2-tailed Student t test. (C) Dami cells knocked down for PTPRJ (PTPRJ KO) or transduced with a scramble shRNA were treated for 48 hours with thrombopoietin and phorbol 12-myristate 13-acetate to induce maturation toward the megakaryocytic lineage. Maturation was then quantified as the increase in surface expression of the megakaryocyte-specific markers CD61, CD41, and CD42b with respect to untreated cells (ie, cells incubated in parallel with medium alone). Surface expression of each antigen was evaluated by flow cytometry as the mean fluorescence intensity (MFI). PTPRJ-KO Dami cells exhibited a similar increase in CD61 and CD41 expression compared with control cells but a defective increase in the late megakaryocyte maturation marker CD42b. Data represent the mean ± SD of 3 independent experiments. *P < .05, 2-tailed Student t test.
PTPRJ knockdown results in inhibition of SDF1-driven migration of the megakaryocytic Dami cell line, as well as in reduced thrombopoietin and phorbol-induced maturation of Dami cells. Using lentiviral transduction of a pool of shRNA, we generated Dami cells expressing <10% PTPRJ protein (PTPRJ KO) compared with cells transduced with the negative control vector. (A-B) SDF1-driven migration of Dami cells was investigated using a Transwell assay. Transwell systems having a polycarbonate membrane with an 8-µm pore size were coated with fibrinogen (A) or type I collagen (B). Aliquots of 1 × 105 cells were seeded in the upper chamber of the Transwell system, whereas medium with 300 ng/mL SDF1 (+) or without SDF1 (–) (negative control conditions) was added to the lower chamber. After incubation for 16 hours at 37°C and 5% CO2, the percentage of cells that migrated to the lower chamber with respect to the number of cells put in the upper chamber at the beginning of the experiment was calculated. Each experiment was performed in duplicate, and data represent the mean of 3 separate experiments. *P < .01, 2-tailed Student t test. (C) Dami cells knocked down for PTPRJ (PTPRJ KO) or transduced with a scramble shRNA were treated for 48 hours with thrombopoietin and phorbol 12-myristate 13-acetate to induce maturation toward the megakaryocytic lineage. Maturation was then quantified as the increase in surface expression of the megakaryocyte-specific markers CD61, CD41, and CD42b with respect to untreated cells (ie, cells incubated in parallel with medium alone). Surface expression of each antigen was evaluated by flow cytometry as the mean fluorescence intensity (MFI). PTPRJ-KO Dami cells exhibited a similar increase in CD61 and CD41 expression compared with control cells but a defective increase in the late megakaryocyte maturation marker CD42b. Data represent the mean ± SD of 3 independent experiments. *P < .05, 2-tailed Student t test.
PTPRC expression
Some reports indicated that human platelets express PTPRC, although to a lesser extent than PTPRJ (www.plateletomics.com).38 Like PTPRJ, PTPRC is a receptor-like tyrosine phosphatase, which, in leukocytes, may exert the same activities as PTPRJ.39,40 To further characterize the consequences of the PTPRJ deficiency, we investigated PTPRC expression in probands’ platelets and megakaryocytes. Flow cytometry did not identify detectable levels of PTPRC in the platelet membrane in patients or in HCs (supplemental Figure 9A). Immunoblotting identified a PTPRC band in platelets; PTPRC expression in the 2 probands was similar to that of healthy subjects (supplemental Figure 9B-C). Cultured CD41+ megakaryocytes were found to express PTPRC, and the expression levels detected in the patients were similar to those of HCs (supplemental Figure 9D-E).
Discussion
Phosphorylation of protein tyrosine residues is one of the main ways by which activation signals are transmitted in cells. Protein tyrosine phosphorylation is controlled by the combined activity of protein tyrosine kinases and protein tyrosine phosphatases (PTPs). The classical PTPs include receptor-type PTPs (RPTPs) and intracellular PTPs, depending on whether they have a transmembrane domain.41 PTPRJ is an RPTP expressed in several cells types, including hematopoietic cells, endothelial cells, fibroblasts, smooth muscle cells, thyroid cells, and mammary cells. Its structure consists of a single intracellular catalytic PTP domain, the transmembrane domain, and an extracellular domain composed of 9 fibronectin type III–like repeats.41,42 In hematopoietic tissue, the most abundant RPTPs are PTPRC and PTPRJ. Although leukocytes contain high levels of PTPRC and, to a lesser extent, PTPRJ, PTPRJ is the most abundant RPTP in platelets and megakaryocytes. Human platelets express ∼2800 copies of PTPRJ on their surface, with little variation among different individuals.24 A series of studies on genetically modified mice showed that PTPRJ is a master regulator of the activity of SFKs in platelets and megakaryocytes, and, as such, has a central role in controlling the transmission of signals in these cells. In particular, PTPRJ acts primarily as an activator of SFKs, although some evidence indicated that, in some conditions, it may also inhibit SFKs activity.24,25,43,44
Here, we have shown that the almost complete loss of PTPRJ due to biallelic loss-of-function mutations in the encoding gene causes a novel form of IT that is transmitted as a recessive trait. In the exomes of the 2 analyzed probands, the PTPRJ-null mutations were the only variants inherited from each healthy parent. The candidacy of this locus was supported by a series of in vitro, in vivo, and ex vivo studies. To investigate the causative role of the loss of PTPRJ on thrombocytopenia, we targeted its functional ortholog gene in vivo in an established zebrafish model.18 CRISPR/Cas9-mediated ablation of ptprja led to a significant and reproducible depletion of CD41+ thrombocytes, the equivalent of platelets in zebrafish. Moreover, silencing of PTPRJ in a human megakaryocytic cell line reproduced 2 functional defects observed in patients’ megakaryocytes that can underlie defective platelet production (ie, impaired SDF1-driven migration and reduced maturation), further supporting the pathogenic role of the PTPRJ-null variants.
Despite PTPRJ being expressed in many cells and tissues, patients with loss of PTPRJ expression did not show phenotypes other than the platelet defect. Unlike the most frequent forms of IT, which are characterized by increased platelet size,2 our patients showed a substantial proportion of small platelets upon examination of blood films. Platelet morphology did not present other obvious alterations. Thrombocytopenia was associated with defective platelet responses to collagen and convulxin, and, to a lesser extent, TRAP, which were evident in the aggregation assay and as impaired P-selectin translocation to plasma membrane and GPIIb-IIIa activation. These functional defects resemble those observed in a Ptprj-knockout mouse model,24 suggesting that they represent a consistent feature of the loss of this phosphatase. Investigation of these mice indicated that the impaired GPVI signaling is likely explained by reduced activation of SFKs in resting platelets. In fact, Ptprj plays a key role in activating SFKs in mouse platelets, because it binds and dephosphorylates their C-terminal tail inhibitory tyrosine residues, thus allowing the conformational changes that lead to autophosphorylation of the activation tyrosine residues.24,44,45 In T and B lymphocytes and macrophages, this action is achieved by Ptprc and Ptprj,39,40 whereas in platelets, Ptprj is the main phosphatase that positively regulates SFKs.43 Through this mechanism, Ptprj maintains a pool of active SFKs in mouse resting platelets, which is essential for the physiologic response to GPVI stimulation by collagen or convulxin.24,25 In line with this model, we observed markedly reduced activation of the SFK SRC in resting and activated platelets from proband II-2, suggesting that all of these mechanisms are also operative in human platelets.
The other forms of IT that may present as a recessive and nonsyndromic thrombocytopenia with normal or reduced platelet size are congenital amegakaryocytic thrombocytopenia caused by MPL or THPO mutations2,46,47 and X-linked thrombocytopenia due to mutations in WAS.48 Thus, once the genetic origin of thrombocytopenia is recognized, the differential diagnosis should consider these 2 disorders together with the IT caused by PTPRJ mutations.
Investigation of megakaryocytes from the patients carrying PTPRJ variants revealed 3 functional alterations that can underlie defective platelet production. As they mature, megakaryocytes migrate in the BM from the osteoblastic niche to areas enriched in sinusoids, where they protrude proplatelets through the vascular endothelium to release them into the circulation. Migration is promoted by a gradient of SDF1, whose concentration is maximal around the sinusoids.27,28,49 Therefore, SDF1-driven migration is a fundamental process for platelet production, and defects in this mechanism have been associated with pathogenesis of some forms of IT, such as those caused by mutations in WAS or MYH9.12,50 Megakaryocytes from patients with PTPRJ mutations showed a substantial reduction in their ability to migrate toward an SDF1 gradient on type I collagen and fibrinogen, 2 of the most abundant proteins in the BM ECM.30,31 It has been previously shown that SFKs have a critical role in motility and migration of megakaryocytes51 ; thus, we hypothesize that this defect is also a consequence of the reduced activation status of these protein tyrosine kinases. Megakaryocytes from Ptprj-knockdown mice exhibited a similar defect in SDF1-driven migration that was associated with defective platelet production. In fact, although these mice did not exhibit overt thrombocytopenia, they showed a significantly reduced rate of platelet recovery after antibody-induced thrombocytopenia.24,44 In addition, megakaryocytes from our patients showed defective proplatelet formation. In fact, we observed a reduced proportion of megakaryocytes extending proplatelets and an alteration in the proplatelet architecture consisting of reduced branching of proplatelet shafts. The latter defect can be directly linked to impaired platelet production, because proplatelet branching serves to multiply the number of proplatelet free ends, the structures that generate platelets. Impaired proplatelet branching is involved in the pathogenesis of several other forms of IT.14,16,52 Reduced activation of SFKs could also contribute to the defective proplatelet formation, since pharmacological inhibition of these kinases inhibited proplatelet extension by human megakaryocytes.53 Thus, the reduced activation status of SFKs due to PTPRJ deficiency appears to be a key mechanism underlying most of the platelet and megakaryocyte defects observed in these patients, including impaired platelet GPVI signaling and defective megakaryocyte migration and proplatelet formation. Finally, patients with PTPRJ mutations presented a reduced in vitro development of CD41+/CD42b+ cells, consistent with a defect in late megakaryocyte maturation, which, in turn, could contribute to the reduced propensity to extend proplatelets. However, the extent of this maturation defect was mild; therefore, its contribution to the pathogenesis of thrombocytopenia is uncertain.
Our data and previous findings suggest that PTPRC is expressed in megakaryocytes and, although at low levels, in platelets.38 PTPRC activity may partially attenuate the effects of PTPRJ deficiency in humans, even if this function is not sufficient to fully compensate for the effects of PTPRJ loss on platelet production and function. However, our findings excluded the hypothesis of a compensatory upregulation of PTPRC in patients’ cells resulting from the PTPRJ deficiency.
In conclusion, we discovered a new form of hereditary thrombocytopenia that highlights a hitherto unknown and fundamental role for PTPRJ in platelet biogenesis in humans.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Antonia Moretta and Margherita Massa for technical assistance with the flow cytometry analysis.
This work was supported by grants from the IRCCS Policlinico San Matteo Foundation, the IRCCS Burlo Garofolo, and the Cariplo Foundation (Italy; grant 2013-0717).
Authorship
Contribution: C.M. designed and performed research, analyzed and interpreted data, and wrote the manuscript; C.A.D.B., K.L., and S.B. designed research, performed research, and analyzed and interpreted data; M.F., V.B., F.P., and S.M. performed research and analyzed and interpreted data; G.L. and P.G. collected patients’ clinical data and analyzed and interpreted data; P.N. designed research, collected patients’ clinical data, and analyzed and interpreted data; C.L.B., A.S., A.B., and T.P. designed research and analyzed and interpreted data; M.S., N.K., and A.P. designed research, analyzed and interpreted data, and wrote the manuscript; and all authors critically revised the manuscript and approved the final version.
Conflict-of-interest disclosure: N.K. and S.M. are paid consultants for Rescindo Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Marco Seri, Department of Medical and Surgical Sciences, University of Bologna, Via Massarenti 9, 40138 Bologna, Italy; e-mail: marco.seri@unibo.it; and Alessandro Pecci, Department of Internal Medicine, IRCCS Policlinico San Matteo Foundation and University of Pavia, Piazzale Golgi, 27100 Pavia, Italy; e-mail: alessandro.pecci@unipv.it.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal