

Key Points
CXCL1 regulates neutrophil homeostasis after pulmonary bacterial pneumonia-induced sepsis.
CXCL2 and CXCL5, after infection rescues, impaired neutrophil-dependent host defense in Cxcl1−/− mice after bacterial pneumonia-induced sepsis.

Abstract
Neutrophil migration to the site of bacterial infection is a critical step in host defense. Exclusively produced in the bone marrow, neutrophil release into the blood is tightly controlled. Although the chemokine CXCL1 induces neutrophil influx during bacterial infections, its role in regulating neutrophil recruitment, granulopoiesis, and neutrophil mobilization in response to lung infection-induced sepsis is unclear. Here, we used a murine model of intrapulmonary Streptococcus pneumoniae infection to investigate the role of CXCL1 in host defense, granulopoiesis, and neutrophil mobilization. Our results demonstrate that CXCL1 augments neutrophil influx to control bacterial growth in the lungs, as well as bacterial dissemination, resulting in improved host survival. This was shown in Cxcl1−/− mice, which exhibited defective amplification of early neutrophil precursors in granulocytic compartments, and CD62L- and CD49d-dependent neutrophil release from the marrow. Administration of recombinant CXCL2 and CXCL5 after infection rescues the impairments in neutrophil-dependent host defense in Cxcl1−/− mice. Taken together, these findings identify CXCL1 as a central player in host defense, granulopoiesis, and mobilization of neutrophils during Gram-positive bacterial pneumonia-induced sepsis.
Introduction
The accumulation of neutrophils is crucial for the innate immune response against a broad array of infectious agents.1,2 Although neutrophils are critical for clearance of pathogens, excessive recruitment can induce substantial tissue/organ damage.2,3 Therefore, neutrophil homeostasis is critical and 2 separate programs for the generation of neutrophils occur in the bone marrow. The maintenance of a stable basal level of mature neutrophils (granulocytes) in bone marrow occurs through steady-state granulopoiesis, whereas large numbers of neutrophils are generated through emergency granulopoiesis after infection or injury of the host.4 These 2 programs of granulopoiesis are differentially regulated at the transcriptional level.4,5 Mature neutrophils from bone marrow are rapidly mobilized into the blood to migrate to infected tissues. Neutrophil recruitment to tissues is a complex, multistep process tightly regulated by cytokines/chemokines6,7 and transcription factors8,9 as well as through interactions between lectins and integrins.10
ELR (Glu-Leu-Arg motif)+CXC chemokines are produced locally and initiate neutrophil recruitment during inflammation. CXCL1/keratinocyte-derived chemokine (KC), also known as GRO-α in humans, is a prototypic ELR+CXC chemokine and mediates neutrophil recruitment through the CXCR2 receptor. CXCL2/Macrophage Inhibitory protein-2 (MIP2) and CXCL5/LPS-induced chemokine are 2 other ELR+CXC chemokines that act through CXCR2. Mice overexpressing CXCL1/KC in lungs have augmented neutrophil influx, whereas blockade of this chemokine with neutralizing antibodies markedly blunts neutrophil recruitment.11 We and others have shown that CXCL1 orchestrates neutrophil-dependent immunity in Klebsiella pneumoniae– and Aspergillus fumigatus–induced lung inflammation, sepsis, and colitis.12-16 In addition to infectious inflammation, CXCL1 also plays an important role in angiogenesis, tumorigenesis, and wound healing.17,18 These studies shed light on the role of CXCL1 in host defense and homeostasis.
Streptococcus pneumoniae is a significant human pathogen, causing a wide range of diseases including pneumonia, meningitis, and septicemia. Among several reported pneumococcal serotypes (STs), pneumonia caused by ST3 strains are the most common and are associated with higher risk for deaths in adults.19 Similar to ST3 strains, ST2 frequently causes pneumococcal diseases, but these infections are associated with lower mortality.19,20 Using a model of Gram-positive bacterial pulmonary infection, we investigated the role of CXCL1 signaling in neutrophil influx, bacterial clearance, and host survival in pneumococcal pneumonia-derived sepsis. The results indicate that CXCL1 is involved in controlling bacterial infections in the lungs, as well as proliferation and differentiation of neutrophilic granulocytes and their mobilization from bone marrow. Moreover, CXCL1-mediated neutrophil mobilization is CD62L- and CD49d-dependent. Our results reveal a previously unrecognized link through CXCL1 between host defense, granulopoiesis, and neutrophil mobilization from the bone marrow after bacterial lung infection. Furthermore, these findings are of potentially high clinical relevance for a broad spectrum of infection-related pulmonary diseases.
Methods
Mice
Cxcl1−/−, Myd88/Trif−/−, and Nlrc4−/− mice were generated as described previously.14,21,22 Asc−/−, Nlrp3−/−, and Nlrp6−/− mice were obtained from Millennium Pharmaceuticals. All mice were backcrossed 8 to 10 times on the C57BL/6J genetic background. Age- and sex-matched C57BL/6J mice were used as controls. A/J and BALB/cJ, were purchased from Jackson Laboratory. The Institutional Animal Care and Use Committee of the Louisiana State University approved in vivo experiments.
Pneumonia-derived sepsis model
Streptococcus pneumoniae 6303 (ATCC), WU2, A66.1, and D39 strains (gifts from David E. Briles, University of Alabama at Birmingham, AL) were used to induce pneumonia as described.12 All strains were grown overnight at 37°C and 5% CO2 in Todd-Hewitt broth plus 0.5% yeast extract, and then subcultured for 6 to 8 hours to reach the midlogarithmic phase to prepare inoculum. Mice were anesthetized and inoculated intratracheally with S pneumoniae as follows: 5 × 104 colony-forming units (CFU; 6303), 5 × 107 CFU (WU2), 2 × 105 CFUs (A66.1), and 5 × 104 CFU (D39) to induce pneumonia or survival studies.
BALF collection, cell counts, and bacterial burden
Bronchoalveolar lavage fluid (BALF) and organs were collected as described earlier.23,24 In brief, mice were euthanized, trachea was exposed and cannulated with 20-gauge catheter, and the lungs were flushed 4 times with 0.8 mL phosphate-buffered saline (PBS) containing heparin and dextrose. Total and differential cell counts were performed on BALF, using light microscopy. The lungs were excised, homogenized in PBS, and plated in serial dilutions on tryptic soy agar plates (with 5% sheep’s blood) to enumerate the bacterial burden.
Antibodies and reagents
Mice were treated with 50 μg anti-CXCL1 monoclonal antibody (mAb; clone 48415; Thermo Fisher Scientific), anti-CXCL2 polyclonal Ab, or anti-CXCL5 mAb (clone 61905) (R&D Systems) intraperitoneally (i.p.) at 24 and 2 hours before infection,25,26 1 μg murine recombinant CXCL2 or CXCL5 (R&D Systems) intratracheally 1 hour postinfection,27 150 μg L-selectin sheddase inhibitor (TAPI-O or KD-IX-73-4; Sigma Aldrich) i.p. at 0 and 24 hours postinfection,28 and 150 μg anti CD49d mAb (BioXCell) i.p. at 0 and 24 hours postinfection.29 Control mice received PBS or dimethyl sulfoxide or control rat immunoglobulin G (IgG), as appropriate.
Flow cytometry
Single-cell suspensions were prepared from lungs, bone marrow, and blood. The femur/tibia were flushed thoroughly with PBS (with 1 mM EDTA) to completely retrieve bone marrow (BM) cells, red blood cells were lysed, and then cells were stained. Lung digestion was performed as described previously.30 All antibodies were purchased from BioLegend unless otherwise listed. For immunophenotyping, the following antibodies were used: anti-CD45 (clone; 30-F11, eBioscience), CD11b (M1/70), Ly6G (1A8), F-4/80 (BM8), CD115 (AFS98), CD3 (17A2), CD4 (GK1.5), CD8α (53-6.7), γδ-TCR (GL3), and NK1.1 (PK136). For studying granulopoiesis,8 the following antibodies were used: anti-Ly6G (IA8), c-Kit (2B8), CD34 (RAM34; eBioscience), and the cocktail of CD3ε (145-2C11), CD4 (RM4.5), CD19 (6D5), CD8α (53-6.7), B220 (RA3-6B2), and TER119 (Ter119). Check supplemental Methods, available on the Blood Web site, for the detailed fluorescence-activated cell sorter (FACS) protocol on granulopoiesis. For hematopoietic stem cells (HSCs) and myeloid progenitors,5,8 BM cells were stained with anti-CD34 (RAM34; eBioscience), FCRγIII/II (93), Sca-1 (D7), c-Kit (2B8), and lineage cocktail (Lin-1). The following antibodies were used to stain adhesion molecules: Ly6G (1A8), CD11b (M1/70), CD11a (121/7), CD29 (HMb1-1), CD18 (GAME-40; BD Biosciences), CD49d (R1-2), and CD62L (MEL-14). Appropriate isotype controls were used for compensation. Cells were acquired either on FACS Calibur or LSRFortessaX20 (BD Biosciences). FlowJo 10 (Treestar) was used to analyze data.
Bone marrow chimeras
Statistics
Prism 7.0a software (GraphPad Software Inc.) was used for statistical analysis. Unpaired Student t test or Mann-Whitney U test (nonparametric) or 1-way ANOVA (followed by Bonferroni’s post hoc comparisons) were used to analyze differences between groups, as appropriate. Survival curves (Kaplan-Meier plot) were compared using log-rank tests. Data are expressed as means ± standard error of the mean. Data are representative of at least 3 experiments, but survival and chimeric studies were performed twice. A P value of *P < .05, **P < .01, and ***P < .001 was considered significant.
Results
S pneumoniae-induced CXCL1 production requires activation of TLR and NF-kB pathways
To determine whether CXCL1 is produced during pneumococcal pneumonia-induced sepsis, C57BL/6J, A/J, and BALB/cJ mice were intratracheally infected with S pneumoniae 6303 (5 × 104 CFU), a ST3 clinical isolate commonly used in preclinical studies. At 48 hours postinfection, we observed increased production of CXCL1 in BALF in all mouse strains (Figure 1A). In addition to the 6303 strain, infection with other ST3 strains, such as A66.1 and WU2, also resulted in higher levels of CXCL1 in the BALF of C57BL/6J mice (Figure 1B). However, infection with D39 (ST2) did not result in a substantial induction of CXCL1 when compared with the ST3 strain (Figure 1B).

S pneumoniae-induced CXCL1 production requires activation of TLR and NF-kB signaling. (A) C57BL6/J, A/J and BALB/c mice were inoculated intratracheally with 5 × 104 CFU S pneumoniae 6303. (B) C57BL6/J mice were infected intratracheally with S pneumoniae strain 6303 (5 × 104 CFU), A66.1 (2 × 105 CFU), WU2 (5 × 107 CFU), and D39 (5 × 104 CFU) strains or PBS. (A-B) Mice were euthanized at 48 hours postinfection, and CXCL1 was measured in BALF. (C) BMDMs from WT mice were generated as described in supplemental Methods. BMDMs were pretreated with TLR agonists Pam2CSK4 (500 ng/mL), Pam3CSK4 (500 ng/mL), HKSA (108/mL), HKSP (108/mL), LTA (5 μg/mL), and LPS (500 ng/mL) for 4 hours and infected with S pneumoniae 6303 (MOI 10) for 8 hours. CXCL1 level was measured in the supernatant. (D) BMDMs from WT, Nlrp3−/−, Nlrc4−/−, and Nlrp6−/− were infected with S pneumoniae 6303 (MOI 10) for 8 hours. CXCL1 was measured in supernatant. (E) BMDMs from WT, MyD88/Trif−/−, and Asc−/− were infected with S pneumoniae 6303 (MOI 10) for 8 and 18 hours. CXCL1 was measured in supernatant. (F) WT BMDMs were pretreated with 10 μM of inhibitors of NF-κB, p38 (SB203580), JNK (SP600125), ERK (PD098059), or dimethyl sulfoxide and then infected with S pneumoniae 6303 (MOI 10) for 8 hours. CXCL1 was measured in the supernatant. All experiments were performed 3 times. (n = 5-6 mice/infection group; n = 3 mice/control group). In vitro experiments have at least 4 biological replicates. Statistical significance was determined by 1-way ANOVA (followed by Bonferroni’s post hoc comparisons). *P < .05; **P < .01; ***P < .001. HKSA, heat-killed S aureus; LTA, lipoteichoic acid; MOI, multiplicity of infection.
S pneumoniae-induced CXCL1 production requires activation of TLR and NF-kB signaling. (A) C57BL6/J, A/J and BALB/c mice were inoculated intratracheally with 5 × 104 CFU S pneumoniae 6303. (B) C57BL6/J mice were infected intratracheally with S pneumoniae strain 6303 (5 × 104 CFU), A66.1 (2 × 105 CFU), WU2 (5 × 107 CFU), and D39 (5 × 104 CFU) strains or PBS. (A-B) Mice were euthanized at 48 hours postinfection, and CXCL1 was measured in BALF. (C) BMDMs from WT mice were generated as described in supplemental Methods. BMDMs were pretreated with TLR agonists Pam2CSK4 (500 ng/mL), Pam3CSK4 (500 ng/mL), HKSA (108/mL), HKSP (108/mL), LTA (5 μg/mL), and LPS (500 ng/mL) for 4 hours and infected with S pneumoniae 6303 (MOI 10) for 8 hours. CXCL1 level was measured in the supernatant. (D) BMDMs from WT, Nlrp3−/−, Nlrc4−/−, and Nlrp6−/− were infected with S pneumoniae 6303 (MOI 10) for 8 hours. CXCL1 was measured in supernatant. (E) BMDMs from WT, MyD88/Trif−/−, and Asc−/− were infected with S pneumoniae 6303 (MOI 10) for 8 and 18 hours. CXCL1 was measured in supernatant. (F) WT BMDMs were pretreated with 10 μM of inhibitors of NF-κB, p38 (SB203580), JNK (SP600125), ERK (PD098059), or dimethyl sulfoxide and then infected with S pneumoniae 6303 (MOI 10) for 8 hours. CXCL1 was measured in the supernatant. All experiments were performed 3 times. (n = 5-6 mice/infection group; n = 3 mice/control group). In vitro experiments have at least 4 biological replicates. Statistical significance was determined by 1-way ANOVA (followed by Bonferroni’s post hoc comparisons). *P < .05; **P < .01; ***P < .001. HKSA, heat-killed S aureus; LTA, lipoteichoic acid; MOI, multiplicity of infection.
Because toll-like receptors (TLRs) and nod-like receptors (NLRs) are important for S pneumoniae-elicited pro-inflammatory responses,32,33 we primed BM-derived macrophages (BMDMs) with known TLR ligands and infected these cells with S pneumoniae 6303. BMDMs infected with S pneumoniae 6303 produced abundant CXCL1 in a dose- and time-dependent manner, even in the absence of priming (supplemental Figure 1A). BMDMs stimulated with TLR agonists Pam2CSK4 (TLR2/6), Pam3CSK4 (TLR3/9), HKSA (heat-killed S aureus; TLR2/6), HKSP (heat-killed S pneumoniae; TLR2), LTA (lipoteichoic acid; TLR2), and LPS (TLR4) produced even higher levels of CXCL1 after infection compared with the PBS-treated group (Figure 1C). Furthermore, BMDMs from Nlrp3−/− and Nlrc4−/−, but not Nlrp6−/−, mice produced higher amounts of CXCL1 after infection (Figure 1D). BMDMs from Asc−/− mice also produced higher amounts of CXCL1 (Figure 1E). After S pneumoniae infection, BMDMs from MyD88/Trif−/− mice produced markedly less CXCL1 compared with WT-BMDM, further confirming the importance of TLR signaling in CXCL1 induction (Figure 1E). Pneumococci are known to activate MAPK/NF-κB signaling, which is implicated in the transcription of cytokines and chemokines.34,35 Therefore, we used inhibitors of NF-κB and MAPK pathways to examine their contribution to CXCL1 production during S pneumoniae infection. NF-kB inhibition abrogated the S pneumoniae-induced CXCL1 production (Figure 1F). Surprisingly, blockade of p38 and JNK, but not ERK, markedly enhanced the CXCL1 production (Figure 1F). Taken together, our data indicate S pneumoniae-induced CXCL1 production requires TLR and NF-κB signaling.
CXCL1 controls neutrophil recruitment, bacterial clearance, and survival in pneumococcal pneumonia-derived sepsis
To elucidate the role of CXCL1 in host defense, we infected Cxcl1 gene-deficient (Cxcl1−/−) and WT mice intratracheally with 3 commonly used ST3 S pneumoniae strains: 5 × 104 (6303), 2 × 105 (A66.1), and 5 × 107 (WU2) CFU. As anticipated, Cxcl1−/− mice displayed attenuated neutrophil recruitment and impaired bacterial clearance in BALF, lungs, and spleen, which was associated with increased mortality after 6303 infection (Figure 2A-C). In addition, CXCL1 differentially regulates recruitment and homeostasis of other immune cells, such as macrophages/monocytes and CD8 T cells (supplemental Figure 2A). Similar to the survival observed after 6303 infection, Cxcl1−/− mice infected with A66.1 and WU2 exhibited similar defects in neutrophil accumulation, bacterial clearance, and host survival (Figure 2D-E; supplemental Figure 3A-B). Mice were also infected with ST2 (5 × 104 CFU; D39) to examine whether the phenotype observed is ST3-specific. However, D39-infected Cxcl1−/− mice displayed similar neutrophil recruitment and bacterial burden compared with WT mice (supplemental Figure 3C). Furthermore, to determine whether the protective role of CXCL1 was mouse strain-specific, we used anti-CXCL1 mAb to neutralize CXCL1 activity in mice of C57BL/6J, A/J, and BALB/cJ backgrounds. As anticipated, anti-CXCL1–treated mice showed reduced neutrophil influx and bacterial clearance in all 3 backgrounds after S pneumoniae 6303 infection (Figure 2F-G).

CXCL1 controls neutrophil recruitment, bacterial clearance, and survival in pneumococcal lung infection. WT and Cxcl1−/− mice were infected intratracheally with S pneumoniae 6303 (5 × 104 CFU; A-C), A66.1 (2 × 105 CFU; D), WU2 (5 × 107 CFU; E) strains or PBS (control). (A) Total number of neutrophils in BALF. (B) The bacterial burden in the BALF and spleen was quantitated at 48 hours postinfection. (C-E) Survival was monitored up to 8 days. A Kaplan-Meier plot is used to show survival of mice from each group. (F-G) C57BL/6J, A/J, and BALB/cJ mice were treated with anti-CXCL1/KC mAb or IgG i.p. at 24 and 2 hours before infection and infected intratracheally with S pneumoniae 6303 (5 × 104 CFU). (F) Neutrophil counts in BALF were measured through the diff-quick method. (G) The bacterial burden in the lungs was quantitated. (H-J) Bone marrow reconstituted mice were infected intratracheally with S pneumoniae 6303 (5 × 104 CFU). Neutrophil count in BALF (H) and bacterial burden in BALF (I) and lungs (J) were quantitated at 48 hours postinfection (n = 5-6 mice/infection group; n = 10 mice/survival; n = 3 mice/control group). All experiments were performed 3 times, with the exception of the survival and chimera studies, which were performed twice. Statistical significance was determined by unpaired t-test (A,F), Mann-Whitney (B,G), log-rank (C-E), and 1-way ANOVA (followed by Bonferroni’s post hoc comparisons; H-J). *P < .05; **P < .01.
CXCL1 controls neutrophil recruitment, bacterial clearance, and survival in pneumococcal lung infection. WT and Cxcl1−/− mice were infected intratracheally with S pneumoniae 6303 (5 × 104 CFU; A-C), A66.1 (2 × 105 CFU; D), WU2 (5 × 107 CFU; E) strains or PBS (control). (A) Total number of neutrophils in BALF. (B) The bacterial burden in the BALF and spleen was quantitated at 48 hours postinfection. (C-E) Survival was monitored up to 8 days. A Kaplan-Meier plot is used to show survival of mice from each group. (F-G) C57BL/6J, A/J, and BALB/cJ mice were treated with anti-CXCL1/KC mAb or IgG i.p. at 24 and 2 hours before infection and infected intratracheally with S pneumoniae 6303 (5 × 104 CFU). (F) Neutrophil counts in BALF were measured through the diff-quick method. (G) The bacterial burden in the lungs was quantitated. (H-J) Bone marrow reconstituted mice were infected intratracheally with S pneumoniae 6303 (5 × 104 CFU). Neutrophil count in BALF (H) and bacterial burden in BALF (I) and lungs (J) were quantitated at 48 hours postinfection (n = 5-6 mice/infection group; n = 10 mice/survival; n = 3 mice/control group). All experiments were performed 3 times, with the exception of the survival and chimera studies, which were performed twice. Statistical significance was determined by unpaired t-test (A,F), Mann-Whitney (B,G), log-rank (C-E), and 1-way ANOVA (followed by Bonferroni’s post hoc comparisons; H-J). *P < .05; **P < .01.
Both hematopoietic and nonhematopoietic cells produce CXCL1 during bacterial infection.12 To study the relative contribution of compartments, we reconstituted lethally irradiated WT or Cxcl1−/− mice with bone marrow from either WT or Cxcl1−/− mice to generate 4 chimeric mouse groups: WT→WT, WT→Cxcl1−/−, Cxcl1−/−→WT, and Cxcl1−/−→ Cxcl1−/−. Compared with the WT→WT group, chimeric groups harboring CXCL1 deficiency in hematopoietic cells (Cxcl1−/−→WT), nonhematopoietic (WT→Cxcl1−/−), and both (Cxcl1−/−→Cxcl1−/−) compartments exhibited defective neutrophil recruitment (Figure 2H). In addition, the expression of CXCL1 in the hematopoietic compartment (WT→Cxcl1−/−) was critical for neutrophil accumulation and bacterial clearance, as the Cxcl1−/−→Cxcl1−/− group had a higher bacterial burden associated with decreased neutrophil influx (Figure 2H-I). Similarly, the loss of CXCL1 in the hematopoietic compartment (Cxcl1−/−→WT and Cxcl1−/−→Cxcl1−/−) resulted in higher bacterial burdens in BALF and lungs compared with the WT→Cxcl1−/− group (Figure 2I-J). However, the chimeric (Cxcl1−/−→WT) group had a tendency to recruit fewer neutrophils and have more bacterial burden in BALF compared with the (WT→Cxcl1−/−) group (Figure 2H-I). Collectively, these data suggest that hematopoietic compartment-derived CXCL1 might be more important than CXCL1 in the stromal compartment for host defense, such as enhanced neutrophil recruitment and augmented bacterial clearance.
Acute pneumococcal lung infection induces emergency granulopoiesis through CXCL1
Neutrophils are constantly produced in the bone marrow through granulopoiesis, and are released to blood or tissue on demand.4 This process is critical for clearance of S pneumoniae from the lungs.36 Thus, the reduced inflammatory response in the lungs of Cxcl1−/− mice and resulting increased mortality after pneumococcal infection raised the possibility that emergency granulopoiesis may be altered in these mice. To address this question, we examined the granulopoietic compartments, using flow cytometry, as described previously.8 During granulopoiesis, neutrophils lose c-Kit and gradually acquire the Ly6G marker. Similar to Candidemia-induced granulopoiesis,8 bone marrow cells in pneumococcal pneumonia-derived sepsis can be divided into 5 subpopulations (#1-#5), based on their expression of c-Kit and Ly6G (Figure 3A). In granulopoietic compartments, subpopulations #1 (c-KithighLy6Gneg), #2 (c-KitintLy6Gneg), and #3 (c-Kitint-negLy6Gneg-int) represent early granulocytic precursors, whereas subpopulations #4 (c-Kitint-negLy6Gint) and #5 (c-KitnegLy6Ghigh) are immature and mature neutrophils, respectively, ready to be released (Figure 3A). At 48 hours after pneumococcal pneumonia-derived sepsis, WT bone marrow exhibited an increase in subpopulations #1 and #4 and a decrease in subpopulation #5 when compared with steady state control mice (Figure 3B-D). In contrast, S pneumoniae-infected Cxcl1−/− mice did not exhibit any changes in subpopulations #1 and #5, but had an increase in subpopulation #4 during pneumococcal pneumonia-derived sepsis (Figure 3B-D). In fact, the frequency and numbers of cells in subpopulation #5 were greater in Cxcl1−/− mice compared with WT mice after infection (Figure 3B-D). However, the distributions of cells in subpopulations #1 to #5 were identical between WT and Cxcl1−/− mice at steady state (Figure 3B-D). These findings suggest CXCL1 is involved in emergency granulocyte generation, but not steady-state granulopoiesis, during pulmonary pneumococcal infection.

Pneumococcal pneumonia-derived sepsis-induced emergency granulopoiesis requires CXCL1. WT and Cxcl1−/− mice were infected intratracheally with S pneumoniae 6303 strain (5 × 104 CFU). (A) Flow cytometric analysis of granulopoiesis. First, bone marrow cells that had lost the potential to give rise to granulocytes were removed from the target population. The remaining cells (R5) were then analyzed for expression of c-Kit and Ly-6G. Populations R2 and R4 represent eosinophilic and megakaryocyte-erythroid progenitors, respectively. FACS dot plot (B), percentage (C), and number (per femur/tibia) (D) of subpopulations #1 to #5 within the granulopoietic compartment at 48 hours postinfection (n = 5-6 mice/infection group; n = 3 mice/control group). All experiments were performed 3 times. Statistical significance was determined by unpaired Student t test (C-D). *P < .05; **P < .01.
Pneumococcal pneumonia-derived sepsis-induced emergency granulopoiesis requires CXCL1. WT and Cxcl1−/− mice were infected intratracheally with S pneumoniae 6303 strain (5 × 104 CFU). (A) Flow cytometric analysis of granulopoiesis. First, bone marrow cells that had lost the potential to give rise to granulocytes were removed from the target population. The remaining cells (R5) were then analyzed for expression of c-Kit and Ly-6G. Populations R2 and R4 represent eosinophilic and megakaryocyte-erythroid progenitors, respectively. FACS dot plot (B), percentage (C), and number (per femur/tibia) (D) of subpopulations #1 to #5 within the granulopoietic compartment at 48 hours postinfection (n = 5-6 mice/infection group; n = 3 mice/control group). All experiments were performed 3 times. Statistical significance was determined by unpaired Student t test (C-D). *P < .05; **P < .01.
Cxcl1−/− mice have reduced amplification of early granulocyte precursors during pneumococcal lung infection-induced emergency granulopoiesis
Granulocytes are generated from HSCs via common myeloid progenitors, granulocyte/macrophage lineage-restricted progenitors (GMPs), and megakaryocyte-erythroid progenitors.37 Compared with WT mice, Cxcl1−/− mice had a reduction in subpopulation #1 during pneumonia, suggesting a possible defect in the generation of HSCs and early granulocyte precursors (Figure 3). We next assessed the composition of the early granulocytic compartments, particularly HSCs and myeloid progenitors, in WT and Cxcl1−/− mice after pulmonary pneumococcal infection. Both WT and Cxcl1−/− mice showed significant increases in the percentage and total number of HSCs (c-Kit+Sca-1+Lin−) during pneumonia-derived sepsis compared with that at steady state, and the numbers of HSCs were identical in both groups of mice (Figure 4A-C). At 48 hours postpneumococcal infection, the population of GMPs, but not common myeloid progenitors or megakaryocyte-erythroid progenitors, increased within the c-Kit+Sca-1−Lin− HSC subpopulation in both WT and Cxcl1−/− mice (Figure 4D-F). However, the increase in GMPs was significantly higher in WT mice compared with Cxcl1−/− mice after infection (Figure 4D-F). Moreover, the numbers of these myeloid progenitors in WT and Cxcl1−/− mice were identical at steady state (Figure 4D-F). Collectively, these data indicate that CXCL1 is required for proliferation of early granulocyte precursors during pneumococcal pneumonia-induced sepsis.

Cxcl1−/−mice have reduced amplification of early granulocyte precursors in pneumococcal lung infection-induced emergency granulopoiesis. (A-F) WT and Cxcl1−/− mice were infected intratracheally with S pneumoniae 6303 (5 × 104 CFU). FACS analysis plot (A), percentage (B), and number (per femur/tibia) (C) of hematopoietic stem cells ((c-Kit+Sca-1+Lin−) at 48 hours postinfection. FACS dot plot (D), percentage (E), and number (per femur/tibia) (F) of myeloid progenitor cells (c-Kit+Sca-1−Lin−) at 48 hours postinfection (n = 5-6 mice/infection group; n = 3 mice/control group). All experiments were performed 3 times. Statistical significance was determined by unpaired Student t (B-C,E-F). *P < .05; **P < .01.
Cxcl1−/−mice have reduced amplification of early granulocyte precursors in pneumococcal lung infection-induced emergency granulopoiesis. (A-F) WT and Cxcl1−/− mice were infected intratracheally with S pneumoniae 6303 (5 × 104 CFU). FACS analysis plot (A), percentage (B), and number (per femur/tibia) (C) of hematopoietic stem cells ((c-Kit+Sca-1+Lin−) at 48 hours postinfection. FACS dot plot (D), percentage (E), and number (per femur/tibia) (F) of myeloid progenitor cells (c-Kit+Sca-1−Lin−) at 48 hours postinfection (n = 5-6 mice/infection group; n = 3 mice/control group). All experiments were performed 3 times. Statistical significance was determined by unpaired Student t (B-C,E-F). *P < .05; **P < .01.
CXCL1 regulates CD62L- and CD49d-dependent neutrophil mobilization in pneumococcal pneumonia-derived sepsis
Next, we investigated the potential role of CXCL1 in release of neutrophils from marrow. To this end, we examined neutrophil retention in the bone marrow of WT and Cxcl1−/− mice at 48 hours postpneumococcal lung infection. Cxcl1−/− mice retained a significantly higher percentage of CD11b+Ly6G+ neutrophils in the bone marrow during pulmonary pneumococcal infection, suggesting a potential defect in the release of mature neutrophils (Figure 5A). Accordingly, Cxcl1−/− mice also exhibited a decrease in CD11b+Ly6G+ neutrophils in the blood and lungs (Figure 5A) when compared with WT mice. CXCR2 and CXCR4 play dominant but opposing roles in the release of mature neutrophils from bone marrow.38 Disruption of CXCR4, whose major ligand is CXCL12/SDF1-α, results into spontaneous mobilization of neutrophils into blood.39 Therefore, we investigated the expression level of CXCR4 on BM neutrophils from Cxcl1−/− mice during pneumococcal pneumonia-induced sepsis. Surprisingly, the expression of CXCR4 on marrow neutrophils did not differ between the steady state and during pneumonia-induced sepsis (Figure 5B). Similarly, Cxcl1−/− mice also produced comparable levels of CXCL12 in the bone marrow after pneumococcal infection (Figure 5C). These results indicate that the defects in neutrophil mobilization in Cxcl1−/− mice are independent of the CXCR4/CXCL12 retention signal in marrow.

CXCL1 regulates CD62L- and CD49d-dependent neutrophil mobilization in pneumococcal pneumonia-derived sepsis. (A-G) WT and Cxcl1−/− mice were infected intratracheally with S pneumoniae 6303 (5 × 104 CFU). Mice were euthanized at 48 hours postinfection. (A) Neutrophil (CD11b+Ly6G+) counts in bone marrow, blood, and lungs were measured. (B) Mean fluorescence intensity of CXCR4 on bone marrow neutrophil was measured. (C) Level of CXCL12 in bone marrow lysates. (D-E) Representative histograms and mean fluorescence intensities of the CD62L in neutrophils. (F-G) Representative histograms and mean fluorescence intensities of the CD49d in neutrophils. (H-I) WT and Cxcl1−/− mice were infected intratracheally with S pneumoniae 6303 (5 × 104 CFU). Mice were treated with L-selectin sheddase inhibitor (TAPI-O or KD-IX-73-4) or mAb against CD49d or vehicle control (dimethyl sulfoxide or IgG) i.p. at 0 and 24 hours postinfection and then euthanized at 48 hours postinfection. The total number of BM neutrophils (CD11b+Ly6G+) per femur/tibia (H) and the percentage of blood neutrophil (CD11b+Ly6G+) (I) were measured. (n = 5-6 mice/infection group; n = 3 mice/control group). All experiments were performed 3 times. Statistical significance was determined by unpaired Student t test (A-G) and 1-way ANOVA (followed by Bonferroni’s post hoc comparisons; H-I). *P < .05; **P < .01; ***P <.001.
CXCL1 regulates CD62L- and CD49d-dependent neutrophil mobilization in pneumococcal pneumonia-derived sepsis. (A-G) WT and Cxcl1−/− mice were infected intratracheally with S pneumoniae 6303 (5 × 104 CFU). Mice were euthanized at 48 hours postinfection. (A) Neutrophil (CD11b+Ly6G+) counts in bone marrow, blood, and lungs were measured. (B) Mean fluorescence intensity of CXCR4 on bone marrow neutrophil was measured. (C) Level of CXCL12 in bone marrow lysates. (D-E) Representative histograms and mean fluorescence intensities of the CD62L in neutrophils. (F-G) Representative histograms and mean fluorescence intensities of the CD49d in neutrophils. (H-I) WT and Cxcl1−/− mice were infected intratracheally with S pneumoniae 6303 (5 × 104 CFU). Mice were treated with L-selectin sheddase inhibitor (TAPI-O or KD-IX-73-4) or mAb against CD49d or vehicle control (dimethyl sulfoxide or IgG) i.p. at 0 and 24 hours postinfection and then euthanized at 48 hours postinfection. The total number of BM neutrophils (CD11b+Ly6G+) per femur/tibia (H) and the percentage of blood neutrophil (CD11b+Ly6G+) (I) were measured. (n = 5-6 mice/infection group; n = 3 mice/control group). All experiments were performed 3 times. Statistical significance was determined by unpaired Student t test (A-G) and 1-way ANOVA (followed by Bonferroni’s post hoc comparisons; H-I). *P < .05; **P < .01; ***P <.001.
Neutrophils mobilized in response to infectious stimuli or chemokines shed L-selectin (CD62L) and express elevated level of CD18 or CD49d-integrins.6,40 Thus, we set out to assess whether there are changes in the expression of adhesion molecules on bone marrow neutrophils during pneumococcal lung infection. At 48 hours postinfection, WT neutrophils show marked shedding of CD62L, but this process was impaired in neutrophils from Cxcl1−/− mice (Figure 5D-E). In addition to defective shedding of CD62L, neutrophils from Cxcl1−/− mice display decreased expression of CD49d compared with WT cells (Figure 5F-G). However, our results show no change in levels of β2-integrins (CD11b/CD18, CD11a/CD18) or β1-integrins (CD29) in Cxcl1−/− mice during pneumococcal lung infection (supplemental Figure 4A). To further confirm the roles of CD49d and CD62L in our pneumonia-derived sepsis model, we used the L-selectin sheddase inhibitor (TAPI-O) and a specific mAb against CD49d. Pretreatment with TAPI-O effectively inhibited shedding of CD62L by neutrophils (supplemental Figure 4B). This inhibition was accompanied by higher retention of neutrophils in bone marrow, which was associated with attenuated release of neutrophils into the blood of WT mice (Figure 5H-I). The retention of neutrophils after TAPI-O administration was also observed in Cxcl1−/− mice (Figure 5H). Similarly, CD49d blockade resulted in higher neutrophil retention in the bone marrow and reduced mobilization of neutrophils into the blood during pulmonary pneumococcal infection (Figure 5H-I). Taken together, our data indicate that CXCL1 regulates neutrophil mobilization in a CD62L- and CD49d-dependent manner in pneumococcal infection without disrupting the CXCR4/CXCL12 retention signal.
Administration of recombinant CXCL2 and CXCL5 rescues the impaired host defense and neutrophil release in Cxcl1−/− mice during pneumococcal pneumonia-induced sepsis
In addition to CXCL1, 2 other ELR+CXC neutrophil chemokines (CXCL2 and CXCL5) act through the CXCR2 receptor.41 Although neutrophil migration from bone marrow to lungs is reduced, there is not a complete abrogation of neutrophil migration in Cxcl1−/− mice, suggesting involvement of either CXCL2 or CXCL5. Intriguingly, Cxcl1−/− mice displayed decreased production of CXCL2 and CXCL5 in BALF, but not in the bone marrow (supplemental Figure 5A). These data suggest the reduced chemokine gradient is responsible for the low numbers of neutrophils and impaired bacterial clearance in Cxcl1−/− mice. Finally, we assessed whether the defects in host defense and neutrophil release observed in Cxcl1−/− mice can be rescued by these CXC chemokines. To this end, we administered recombinant CXCL2 and CXCL5 1-hour postinfection with pneumococci. Administration of recombinant CXCL2 resulted in increased neutrophil recruitment and bacterial clearance (Figure 6A-C). Furthermore, administration of recombinant CXCL5 enhanced neutrophil recruitment and bacterial clearance in Cxcl1−/− mice during pneumococcal infection (Figure 6D-F). In addition, recombinant CXCL2 also increased release of mature neutrophils (#P5) from bone marrow (Figure 6G-H). To further validate the roles of CXCL2 and CXCL5, WT mice were treated with either anti-CXCL2 or anti-CXCL5 or both antibodies at 24 and 2 hours before infection. Mice receiving anti-CXCL2 or anti-CXCL5 or both had enhanced retention of the P5 population in granulocytic compartment and reduced blood neutrophils compared with that of IgG-treated mice (supplemental Figure 6A-B,E-F). In addition, the neutralization of both CXCL2 and CXCL5, but not single treatment of either anti-CXCL2 or anti-CXCL5, leads to the decreased GMPs frequency (supplemental Figure 6C-D). Taken together, our data suggest CXCL2 and CXCL5 can compensate for the loss of CXCL1 in pneumococcal lung infections.
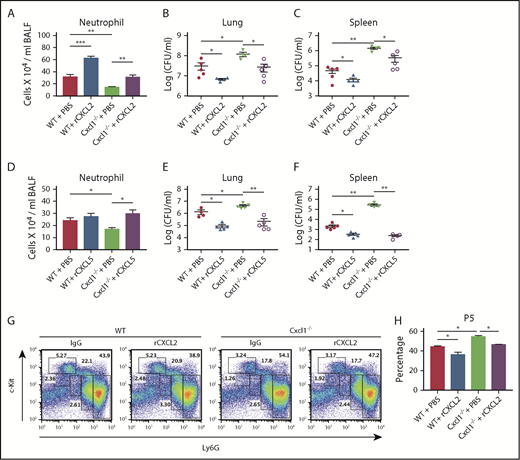
Administration of recombinant CXCL2 and CXCL5 rescues the impaired host defense seen in Cxcl1−/−mice during pneumococcal lung infection. (A-H) WT and Cxcl1−/− mice were infected intratracheally with S pneumoniae 6303 (5 × 104 CFU). Mice were treated with murine recombinant CXCL2 and CXCL5 intratracheally at 1-hour postinfection and then euthanized at 48 hours postinfection. BALF neutrophil counts (A) and bacterial burden in lungs (B) and spleen (C) were quantitated. BALF neutrophil counts (D) and bacterial burden in lungs (E) and spleen (F) were quantitated. FACS dot plot (G) and mature neutrophil (subpopulation #5; Ly6G+c-Kit−) counts in bone marrow (H) were measured. (n = 5-6 mice/infection group; n = 3 mice/control group). All experiments were performed 3 times. Statistical significance was determined by 1-way ANOVA (followed by Bonferroni’s post hoc comparisons; A-H). *P < .05; **P < .01.
Administration of recombinant CXCL2 and CXCL5 rescues the impaired host defense seen in Cxcl1−/−mice during pneumococcal lung infection. (A-H) WT and Cxcl1−/− mice were infected intratracheally with S pneumoniae 6303 (5 × 104 CFU). Mice were treated with murine recombinant CXCL2 and CXCL5 intratracheally at 1-hour postinfection and then euthanized at 48 hours postinfection. BALF neutrophil counts (A) and bacterial burden in lungs (B) and spleen (C) were quantitated. BALF neutrophil counts (D) and bacterial burden in lungs (E) and spleen (F) were quantitated. FACS dot plot (G) and mature neutrophil (subpopulation #5; Ly6G+c-Kit−) counts in bone marrow (H) were measured. (n = 5-6 mice/infection group; n = 3 mice/control group). All experiments were performed 3 times. Statistical significance was determined by 1-way ANOVA (followed by Bonferroni’s post hoc comparisons; A-H). *P < .05; **P < .01.
Discussion
Neutrophil homeostasis is a tightly regulated process that involves granulopoiesis, mobilization, and recruitment to organs/tissues. ELR+CXC chemokines display potent neutrophil chemotactic properties, although their role in neutrophilic granulopoiesis and release from the bone marrow in Gram-positive infections is undefined. Thus, we used a murine model of pneumococcal pneumonia-derived sepsis to investigate the role of CXCL1 in granulopoiesis, and mobilization of neutrophils from the marrow.
A central feature of inflammation is the activation of TLRs, NLRs, transcription factors, and MAP kinases.32-35 Although our results revealed that CXCL1 production is mediated by the TLR-MyD88/Trif-NF-kB axis, the NLR-ASC and MAP kinase pathways might negatively regulate it during pneumococcal infection. These findings suggest that NLR activation may serve as a molecular switch to dampen TLR-dependent pathways and prevent excessive inflammation. Our observations indicate that CXCL1 plays an essential role in pulmonary host defense against ST3 pneumococci (6303, A66, WU2) regulating neutrophil recruitment and bacterial clearance. Using anti-CXCL1 antibody on A/J and BALB/cJ backgrounds, we confirmed that CXCL1-mediated neutrophil-dependent immunity is not background-specific.
The contributions of myeloid cells and stromal cells to neutrophil accumulation in pneumococcal pneumonia-derived sepsis are not known. Myeloid cells produce CXCL1 and CXCL2, and type II alveolar cells produce CXCL5.12,26 Previously, we have shown that CXCL1 derived from both hematopoietic and stromal cells contributes to host defense during Klebsiella infection.12 Here, we report that CXCL1-expressing hematopoietic cells primarily contribute to neutrophil recruitment and bacterial clearance, despite the fact that CXCL1 expression in both compartments is essential for protection against pneumococcal infection.
Although the role of CXCL1 for neutrophil recruitment is well established, its function in granulopoiesis and mobilization from the marrow has not been investigated. We demonstrate that CXCLL1 is important during the amplification of granulocytic progenitor cells in bone marrow. Our data show that CXCL1 is not essential at steady state, but is required for emergency granulopoiesis during pneumococcal infection. In addition, CXCL1 is important for proliferation and differentiation of c-Kit+ early granulocytic precursors into Ly6G+ neutrophils. The increased frequency of granulocytic progenitors in WT mice, but not in Cxcl1−/− mice, could be a result of increased proliferation or cell survival or both of these mechanisms after pneumococcal infection. Moreover, STAT3 and C/EBPβ are known to be master regulators of granulopoiesis in mice.5,8,9 Future studies will be required to investigate whether the reduced granulopoiesis observed in Cxcl1−/− mice is associated with impaired expression of STAT3 or C/EBPβ.
CXCR2, a common receptor for CXC chemokines, is widely known to participate in neutrophil mobilization in response to infections and cytokines.38,41,42 Cxcr2−/− mice have profound defect in neutrophil emigration to sites of inflammation.43 Furthermore, CXCR2 negatively regulates CXCR4-mediated neutrophil retention within the bone marrow.38 CXCR2 engagement induces a heterologous desensitization to the effects of CXCL12 on CXCR4, thereby inducing neutrophilia.44 Because Cxcl1−/− mice have unaltered levels of neutrophil CXCR4 and bone marrow CXCL12, it is possible that the loss of CXCL1-CXCR2 engagement may have compromised CXCR4 desensitization in our model.
Neutrophil mobilization greatly differs between homeostatic and inflammatory settings. Our findings reveal that CXCL1 mediates CD62L- and CD49d-dependent release of neutrophils from the marrow during pneumococcal infection. The bone marrow sinusoidal endothelium constitutively expresses cell adhesion molecules.45 Although maturation-related loss of both CD49d and CXCR4 have been shown to regulate release of neutrophil in the homeostatic setting,46 we believe that massive release of often less mature neutrophils during pneumococcal pneumonia may be much more complex and different under the influence of mediators such as CXCL1. Because we did not see much alteration in marrow CXCL12 and neutrophil CXCR4 expression, we analyzed the panel of adhesion molecules, results of which show marked alteration in expression of CD62L and CD49d only (Figure 5). Similar to our study, MIP2 (CXCR2-ligand) has been shown to regulate CD49d-dependent neutrophil mobilization.6 In contrast, G-CSF-induced neutrophilia is predominantly associated with disruption of CXCR4/CXCL12 signal.47 Thus, we speculate that CD49d-dependent neutrophil mobilization may be an important mechanism when inflammatory response is primarily mediated through CXCR2 engagement and the expression of CXCR4/CXCL12 signal remains unaltered. However, further studies are needed to address the speculation.
Our findings indicate that neutrophils shed CD62L during pneumococcal pneumonia-derived sepsis, and CXCL1 deficiency impairs this process and leads to the increased retention of mature neutrophils in Cxcl1−/− mice. These findings are in support of previous reports showing the relevance of CD62L shedding to neutrophil mobilization.6,48 The loss of CD62L surface expression during inflammatory response is primarily associated with proteolytic cleavage of the ecto-domain of L-selectin by tumor necrosis factor-α converting enzymes, which may be regulated by p38 MAPK and protein kinase C signaling.49,50 Because administration of TAPI-O reduced the mobilization in our model (Figure 5), it is likely that CXCL1 may also regulate proteolytic cleavage of CD62L during pneumococcal lung infection. We speculate that the decreased expression of CD49d in Cxcl1−/− mice might have also occurred through a similar mechanism, which remains yet to be explored.
In conclusion, the current study identifies a critical role of CXCL1 signaling not only in mediating neutrophil recruitment but also in the generation of neutrophilic granulocytes and their mobilization from bone marrow in response to Gram-positive lung infection. The results of this study support the concept that the mechanisms regulating neutrophilic granulopoiesis and mobilization in infectious settings are different from those that regulate these events at steady state. Moreover, future studies will be required to further define the molecular and cellular mechanisms by which CXCL1 regulates neutrophil homeostasis on pulmonary Gram-positive bacterial infection.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Marilyn Dietrich for flow cytometry and Lung Biology Laboratory members for their helpful discussions.
This work was supported by a predoctoral fellowship from National Institutes of Health, National Heart, Lung, and Blood Institute (F31HL137287) (S.P.), a postdoctoral fellowship from American Heart Association (L.J.), a grant from National Institutes of Health, National, Heart, Lung, and Blood Institute (R01HL133336) (S.C.), and grants from National Institutes of Health, National Institute of Allergy and Infectious Diseases (R01AI113720, R21AI133681) and National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL091958) (S.J.).
Authorship
Contribution: S.P. and S.J. conceived and designed experiments; S.P., P.B., L.G., S.B., L.J., J.A.D., J.T.L., and S.C. performed experiments and collected data; and S.P. and S.J. analyzed the data and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for P.B. is Department of Immunology, Harvard Medical School, Boston, MA.
Correspondence: Samithamby Jeyaseelan, Center for Lung Biology and Disease, Department of Pathobiological Sciences, School of Veterinary Medicine, Louisiana State University, Baton Rouge, LA 70803; e-mail: jey@lsu.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal