

Key Points
Maea, but not Vcam1, is required for adult BM macrophage development and EI function in vivo.
Deletion of Maea expressed by macrophages, but not by EBs, impairs BM EI formation.

Abstract
The erythroblastic island (EI), formed by a central macrophage and developing erythroblasts (EBs), was first described decades ago and was recently shown to play an in vivo role in homeostatic and pathological erythropoiesis. The exact molecular mechanisms, however, mediating the interactions between macrophages and EBs remain unclear. Macrophage-EB attacher (Maea) has previously been suggested to mediate homophilic adhesion bounds bridging macrophages and EBs. Maea-deficient mice die perinatally with anemia and defective erythrocyte enucleation, suggesting a critical role in fetal erythropoiesis. Here, we generated conditional knockout mouse models of Maea to assess its cellular and postnatal contributions. Deletion of Maea in macrophages using Csf1r-Cre or CD169-Cre caused severe reductions of bone marrow (BM) macrophages, EBs, and in vivo island formation, whereas its deletion in the erythroid lineage using Epor-Cre had no such phenotype, suggesting a dominant role of Maea in the macrophage for BM erythropoiesis. Interestingly, Maea deletion in spleen macrophages did not alter their numbers or functions. Postnatal Maea deletion using Mx1-Cre or function inhibition using a novel monoclonal antibody also impaired BM erythropoiesis. These results indicate that Maea contributes to adult BM erythropoiesis by regulating the maintenance of macrophages and their interaction with EBs via an as-yet-unidentified EB receptor.
Introduction
Red blood cell (RBC) homeostasis is tightly regulated by balanced production and clearance. Bone marrow (BM) erythroid precursors were first observed several decades ago in tight association with a central macrophage in a structure referred to as an erythroblastic island (EI).1 Macrophages regulate both normal and diseased erythropoiesis, including promotion of erythroid precursor survival and proliferation, iron homeostasis and transfer, and terminal maturation and enucleation.2-5 These activities are promoted by direct interactions between the macrophage and erythroblast (EB)6,7 via several proposed adhesion mechanisms including (macrophage: EB) Vcam1: very-late antigen 4 (VLA-4),8,9 αV: Icam4,10 or macrophage-EB attacher (Maea): Maea,7 CD163,11 and Palladin.12 However, the exact role of these adhesion molecules during in vivo adult erythropoiesis has not been determined.
Among these, Maea was originally identified as an adhesion molecule expressed by both macrophages and EBs, and was suggested to mediate EI formation via its homophilic interactions.7,13 Targeted gene inactivation of Maea caused severe defects in fetal liver erythropoiesis and macrophage development,14 but the perinatal lethality of Maea-null embryos has prevented detailed examination of its function in adult hematopoiesis. Here, we have generated a conditional allele of Maea and determined that Maea plays a critical role in adult BM macrophage development and EI function. Comparative analysis with Vcam1 deletion shows that Maea exerts a dominant role in the EI. Selective deletion of Maea in the macrophage or EB disrupts BM erythropoiesis only when Maea is deleted in the macrophage, suggesting that Maea may not interact by homophilic interactions.
Methods
Animals
Maeafl/fl mice were generated as described in the next section. Vcam1fl/fl mice15 were kindly provided by Thalia Papayannopoulou (University of Washington School of Medicine, Seattle, WA) and backcrossed to a C57BL/6 strain for at least 10 generations. Csf1r-Cre mice16 were a gift from Jeffrey W. Pollard (University of Edinburgh, Edinburgh, United Kingdom) and also backcrossed onto a C57BL/6 background. CD169-Cre knockin mice were previously generated and described.17 Epor-Cre mice18 were kindly provided by Ann Mullally (Dana-Farber/Brigham and Women’s Hospital, Boston, MA). C57BL/6 (CD45.2) and Bl6-Ly5.1 (CD45.1) mice were purchased from Charles River Laboratories (Frederick Cancer Research Center, Frederick, MD)/National Cancer Institute (NCI) or The Jackson Laboratory (B6.SJL-PtprcaPepcb/BoyJ). R26-tdTomato (B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J) and Mx1-Cre (B6.Cg-Tg(Mx1-cre)1Cgn/J) mice were obtained from The Jackson Laboratory. All animals were housed in a specific pathogen-free barrier facility. All experimental procedures were approved by the Animal Care and Use Committee of Albert Einstein College of Medicine. All experiments were performed on mice of both sexes with littermate controls from the same colony between 6 and 12 weeks of age.
Generation of Maeafl/fl mice
The EuMMCR targeting vector PG00141_Z_1_G10 was purchased and electroporated into WW6 embryonic stem (ES) cells. After drug selection, resistant ES cell colonies were picked and screened by Southern blot analysis using 5′ and 3′ external probes (supplemental Figure 1A, available on the Blood Web site). Correctly targeted ES cell clones were injected into C57BL/6 blastocysts and the resulting chimeric mice were bred with C57BL/6 animals to establish the MaeaTargeted line. Once the germline transmission was confirmed, the MaeaTargeted mice were crossed to Rosa26FLP1 mice (B6.129S4-Gt(ROSA)26Sortm1(FLP1)Dym/RainJ; The Jackson Laboratory) to remove the LacZ/Neo cassette and generate the floxed allele Maeafl. Both alleles were then backcrossed onto a C57BL/6 background for at least 5 generations before crossing to Cre strains for functional studies. Genotyping was done by ear-clip genomic DNA polymerase chain reaction using primers F1 + R1 + R2 and F2 + R3 + R4 (supplemental Figure 1A-B). Primer sequences are as follows: F1, gttcagcctcaggattcagg; R1, atgagcaggggacctcaac; R2, aactgatggcgagctcaga; F2, caccagctcaggcagttaca; R3, ccacaacgggttcttctgtt; R4, cgggaagaagtgggattacc.
Antibodies and flow cytometry
Purified goat anti-MAEA polyclonal antibody (I-20) was purchased from Santa Cruz Biotechnology and used at a 1:100 concentration. Conjugated donkey anti-goat immunoglobulin G (IgG) secondary antibodies were obtained from Thermo Fisher Scientific and used at a 1:800 concentration. Fluorochrome-conjugated or biotinylated antibodies against mouse Gr-1 (Ly6C/G; clone RB6-8C5), CD115 (clone AFS98), B220 (clone RA3-6B2), F4/80 (clone BM8), Vcam1 (clone 429), CD11b (clone M1/70), CD45 (clone 30-F11), Ter119 (clone TER-119), CD71 (clone R17217) and CD44 (clone IM7), CD45.1 (clone A20), and CD45.2 (clone 104) were obtained from BioLegend or eBioscience. Femurs were flushed gently with 1 mL of ice-cold PEB (phosphate-buffered saline [PBS]/2 mM EDTA/0.5% bovine serum albumin) buffer through a 1-mL syringe (BD) with a 21G needle (BD) into fluorescence-activated cell sorting (FACS) tubes. Spleens were mashed through a 40-μm filter (BD) into 6-well plates (BD) containing 4 mL of ice-cold PEB. For EB analysis, single-cell suspensions were isolated by harvesting the interface layer from a lympholyte gradient (Cedar Lane Laboratories) according to the manufacturer’s directions. For other analysis, RBCs were lysed by an ammonium chloride solution. 4′,6-Diamidino-2-phenylindole–negative (DAPI−) singlets were analyzed for all live samples unless otherwise specified. Isolation of in vivo–formed EIs was described previously.19 Briefly, BM was flushed gently with Iscove modified Dulbecco medium (Cellgro) containing 3.5% sodium citrate and 20% fetal calf serum solution using an 18G syringe (BD). Cells were then stained and diluted in FACS buffer containing DAPI without centrifugation or disruption until processed for the F4/80+Ter119+ multiplet population. Stained sample suspensions were acquired on an LSR II (BD) and results were analyzed and visualized by FlowJo (TreeStar). For sorting, samples were processed under sterile conditions and sorted on a BD FACSAria.
Generation of MAEA mAb
BALB/c mice were immunized with a keyhole limpet hemocyanin–conjugated MAEA peptide that is part of the extracellular domain (AAQKN IDRET SHVTM VVAEL EKTLS GCPA) and boosted with the same peptide and recombinant MAEA protein (Novus). Hybridomas producing monoclonal antibodies (mAbs) against human MAEA were generated by standard techniques from splenocytes fused to Ag8.653 or NSObcl2 myeloma cells.20 Clone 92 (IgG2a) was firstly selected by an enzyme-linked immunosorbent assay screen as its mAb recognized MAEA peptide/protein, but not human IgG. Subclone 92.25 was further selected and validated by FACS staining of wild-type, but not MaeaCsf1rCre, mouse BM cells due to only 1-aa difference between the human and mouse sequence in the antibody target region. mAbs were then concentrated and purified from concentrated hybridoma supernatant by Amicon Ultra-15 100K filters (Millipore) and NAb Protein A/G Spin kits (Thermo Fisher Scientific).
Complete blood count
Mice were bled ∼25 μL into an Eppendorf tube containing 2 μL of 0.5 M EDTA (Life Technologies) using heparinized microhematocrit capillary tubes (Fisher Brand) under isoflurane anesthesia. Blood was diluted 1/20 in PBS and analyzed on an Advia counter (Siemens).
In vivo clearance of RBCs
Long-term RBC clearance was assayed as previously described.19 Mice were given a single IV NHS sulfo-biotin (Thermo Fisher Scientific) injection (100 mg/kg), and the fraction of biotinylated RBCs was determined weekly from 1 μL of blood. Short-term clearance was assayed by IV injection of 2 × 108 carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled wild-type RBCs and monitored at indicated time points.
Splenectomy
Mice were splenectomized under 100 mg/kg ketamine and 10 mg/kg xylazine anesthesia as previously described21 and allowed to recover for 4 weeks before experiments.
BM transplantation
All recipient mice were lethally irradiated (600 + 600 cGy, at least 3 hours apart) in a Shepherd Mark 1 irradiator. RBC-lysed BM nucleated cells (1 × 106, unless otherwise indicated) were then injected retro-orbitally under isoflurane anesthesia.
Colony-forming assays
Spleen burst forming unit–erythroid was assayed by plating 5 × 105 RBC-lysed splenocytes in MethoCult M3436 (StemCell Technologies) according to the manufacturer’s instructions, and colonies were enumerated on day 10 of culture.
In vivo treatment
For hemolytic anemia induction, mice were injected intraperitoneally with 40 mg/kg body weight phenylhydrazine (PHZ; Sigma-Aldrich) on day 0 and 1 of the experiment. For 5-fluorouracil (5-FU) challenge, a single dose (250 mg/kg body weight) of freshly made 5-FU was given IV to each mouse under isoflurane anesthesia. Mx1-Cre was induced by 3 doses of polyinosinic:polycytidylic acid (Poly I:C; Invivogen) injections intraperitoneally every other day at 5 mg/kg. For antibody treatment, purified MAEA mAb and control IgG2a (BioXcell) were diluted in PBS and injected intraperitoneally at 100 μg daily for 3 weeks.
Cell culture
In vitro terminal differentiation and enucleation of sorted polychromatic EBs (EB-III) was done as previously described.22 Briefly, EB-III were FACS sorted and cultured at <106/mL in differentiation media composed of Iscove modified Dulbecco medium, 10% fetal bovine serum (FBS), 1% bovine serum albumin, 30 ng/mL erythropoietin (Epo; BioLegend), 0.2 mg/mL holotransferrin (Sigma-Aldrich), and 10 μg/mL insulin (Thermo Fisher Scientific) for 48 hours. At the end of the culture, cells were analyzed by FACS after Ter119 and H33342 staining. An in vitro phagocytosis assay using BM-derived macrophages (BMDMs) or spleen-derived macrophages (SPDMs) was slightly modified from previously described.23 BMDMs or SPDMs were isolated by adherence from BM or splenic suspensions in macrophage media (RPMI 1640 with 10% FBS, 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid [HEPES], and 10 ng/mL macrophage colony-stimulating factor) for 7 days.24,25 On day 7, BMDMs or SPDMs were harvested by gentle scraping and plated at 1 × 105 per well in 12-well plates for 24 hours in full macrophage media. The macrophages were then serum-starved for 2 hours before adding 1 × 107 CFSE-labeled RBCs or 4 × 104 CD45.1 BM cells as target. After 2∼3 hours coincubation, nonadherent cells were washed and macrophages were scraped for FACS analysis.
In vitro reconstitution of EIs
In vitro reconstitution of EIs was done as previously described with minor modifications.14 Briefly, BM cells were resuspended in complete media (RPMI 1640/10% FBS) and allowed to attach to glass coverslips in 6-well plates 4 hours to overnight at 37°C in 5% CO2. Adherent macrophages were then stripped and washed by dipping in RPMI 1640 media and then PBS without calcium and magnesium. Next, BM cells devoid of macrophages by the same procedures were used as the source of EBs and added to the striped macrophages in complete media with 10 μg/mL IgG or anti-Maea antibodies. After 30 minutes of incubation, the coverslips were dipped in RPMI 1640 to remove the unbound cells before being stained for F4/80 and Ter119 and mounted for immunofluorescence imaging using standard procedures. Random fields with the presence of macrophages were taken and the number of attached Ter119+ cells was quantified.
Quantification and statistical analysis
In each experiment, each mouse was analyzed as a biological replicate. Data visualization (shown as mean plus or minus standard error of the mean [SEM]) and statistical analysis were performed using Graphpad Prism 7. Unpaired Student t test was used to assess statistical significance when comparing two samples unless otherwise indicated.
Results
Maea is required for adult BM macrophage development and EI niche formation
To examine Maea function in adult erythropoiesis, we generated mice with a floxed allele of Maea, which leads to a frame shift and nonsense-mediated decay of Maea messenger RNA (mRNA) upon Cre-mediated recombination (supplemental Figure 1A-B). Flow cytometry analysis using a MAEA-specific polyclonal antibody revealed that in adult BM mononucleated cells (BMNCs), Maea was highly expressed in the macrophages with minimal expression by monocytes, neutrophils, or B cells (gated as previously described26 ; Figure 1A-B). We initially intercrossed Maeafl/fl mice with a Csf1r-Cre–transgenic line16 to delete Maea in the monocytic-macrophage lineage. Csf1r-Cre; Maeafl/fl animals (henceforth MaeaCsf1r-Cre) were born healthy and fertile, and survived into adulthood at expected Mendelian ratios, by contrast to the perinatal lethality reported in Maea-null mice.14 Efficient MAEA depletion on BM macrophages was confirmed by FACS analysis (Figure 1A-C). The BM of MaeaCsf1r-Cre mice exhibited a slight, but significant, reduction in cellularity (Figure 1D). Further analysis revealed that their BM macrophage numbers represented ∼30% of wild-type levels (Figure 1E). MaeaCsf1r-Cre bones also appeared paler than controls (Figure 2A), suggesting a reduced erythroid content in the marrow. Indeed, the number of BM EBs was markedly reduced in MaeaCsf1r-Cre BM compared with littermate controls (Figure 2B-C). This is likely due to disruption of EI formation because we observed a marked reduction in EIs (∼30% of control levels) formed in vivo in the MaeaCsf1r-Cre BM (Figure 2D). Profiling of the EB maturation status revealed a partial block of differentiation at the polychromatic (EB-III) stage27,28 (Figure 2E). These results support a critical role of Maea in adult BM EI formation and functions.

Maea deletion impairs BM macrophage development. (A) Representative FACS plots of the BMNC gating strategy for each cell population shown in panel B. (B) Representative histograms showing Maea expression on BM leukocytes from control (Ctrl) littermates and MaeaCsf1r-Cre mice. (C) Deletion efficiency of Maea by Csf1r-Cre on BM macrophages (Mϕ) as determined by FACS (n = 8). (D) Quantification of total BM cellularity in control and MaeaCsf1r-Cre mice (n = 8). (E) Quantification showing significant reduction of macrophages in the BM of MaeaCsf1r-Cre mice compared with littermate controls (n = 8). Data are shown as mean plus or minus SEM. **P < .01, ***P < .001, ****P < .0001 by unpaired Student t test. FSC, forward scatter; MFI, mean fluorescence intensity; SSC, side scatter.
Maea deletion impairs BM macrophage development. (A) Representative FACS plots of the BMNC gating strategy for each cell population shown in panel B. (B) Representative histograms showing Maea expression on BM leukocytes from control (Ctrl) littermates and MaeaCsf1r-Cre mice. (C) Deletion efficiency of Maea by Csf1r-Cre on BM macrophages (Mϕ) as determined by FACS (n = 8). (D) Quantification of total BM cellularity in control and MaeaCsf1r-Cre mice (n = 8). (E) Quantification showing significant reduction of macrophages in the BM of MaeaCsf1r-Cre mice compared with littermate controls (n = 8). Data are shown as mean plus or minus SEM. **P < .01, ***P < .001, ****P < .0001 by unpaired Student t test. FSC, forward scatter; MFI, mean fluorescence intensity; SSC, side scatter.
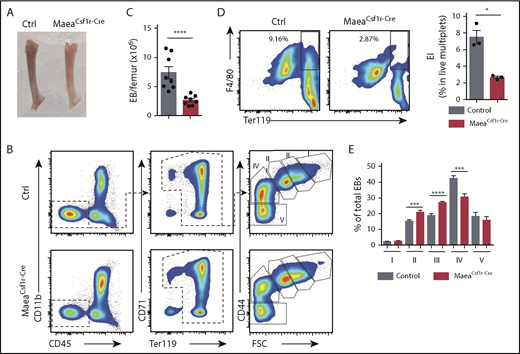
Maea deletion disrupts BM EI niche. (A) Representative photograph of dissected femurs from control and MaeaCsf1r-Cre mice. (B-C) Representative FACS gating strategy and quantification showing significant reduction of BM EBs in MaeaCsf1r-Cre mice (n = 8). (D) Representative FACS plots and quantification showing impaired in vivo formation of BM EIs (F4/80+Ter119+ live multiplets) in MaeaCsf1r-Cre mice (n = 3). (E) Quantification of EBs at various stages of maturation (n = 8) (I, pro-EBs; II, basophilic EBs; III, polychromatic EBs; IV, orthochromatic EBs and reticulocytes; V, mature RBCs). Data are shown as mean plus or minus SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001 by unpaired Student t test.
Maea deletion disrupts BM EI niche. (A) Representative photograph of dissected femurs from control and MaeaCsf1r-Cre mice. (B-C) Representative FACS gating strategy and quantification showing significant reduction of BM EBs in MaeaCsf1r-Cre mice (n = 8). (D) Representative FACS plots and quantification showing impaired in vivo formation of BM EIs (F4/80+Ter119+ live multiplets) in MaeaCsf1r-Cre mice (n = 3). (E) Quantification of EBs at various stages of maturation (n = 8) (I, pro-EBs; II, basophilic EBs; III, polychromatic EBs; IV, orthochromatic EBs and reticulocytes; V, mature RBCs). Data are shown as mean plus or minus SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001 by unpaired Student t test.
Maea is dispensable for RBC enucleation
By contrast to the prior report on Maea-null mice,14 we did not observe peripheral blood anemia in young adult MaeaCsf1r-Cre mice (supplemental Figure 2A). Although a role for Maea in enucleation has also been suggested,14 blood smears and FACS analyses revealed elevated CD71+Ter119+ reticulocyte counts in MaeaCsf1r-Cre animals but no nucleated RBCs in circulating blood (supplemental Figure 2B-D). To further investigate this issue, we sorted polychromatic EBs (EB-III) from BM of MaeaCsf1r-Cre and control mice and evaluated enucleation rates in vitro. We found that cultured MaeaCsf1r-Cre-derived EBs enucleated at a similar rate as those of controls (supplemental Figure 2E). These results suggest that at least macrophage Maea expression is dispensable for postnatal RBC enucleation.
Maea regulates RBC dynamics during stress erythropoiesis
We next challenged control and MaeaCsf1r-Cre mice with hemolytic anemia induced by the hemoglobin-oxidizing reagent PHZ. We observed a significant impairment of the reticulocyte response, but not RBC or hematocrit recovery, in MaeaCsf1r-Cre mice (supplemental Figure 3A). Similarly, reticulocytosis in MaeaCsf1r-Cre mice after a single dose of cytotoxic agent 5-FU was significantly delayed whereas RBC and hematocrit showed an attenuated decline before a mild but significant delay in recovery (supplemental Figure 3B). The attenuated early decline in hematocrit was consistent with previously described macrophage-depleted models,19 suggesting that Maea depletion–caused macrophage defects were contributing to both the RBC production and clearance. Indeed, we found that the RBC lifespan was significantly prolonged in MaeaCsf1r-Cre animals (supplemental Figure 3C). However, we did not detect any phagocytosis defects in MaeaCsf1r-Cre macrophages (supplemental Figure 3D), suggesting the RBC clearance defect likely reflected the overall reduction of macrophage numbers.
Differential roles of Maea in spleen and BM macrophages
Interestingly, even though Csf1r-Cre similarly induced an efficient Maea deletion in splenic red pulp macrophages (RPMs), their numbers were not significantly altered (supplemental Figure 4A; Figure 3A-B), suggesting differential requirements of Maea between BM and spleen macrophage development or maintenance. Spleen EB numbers were not significantly altered, although profiling of their maturation revealed a similar partial block at the polychromatic stage (supplemental Figure 4B-C). However, we found compensatory stress erythropoiesis in the spleen of MaeaCsf1r-Cre mice evidenced by splenomegaly and elevated burst-forming unit–erythroid numbers (supplemental Figure 4D-E).
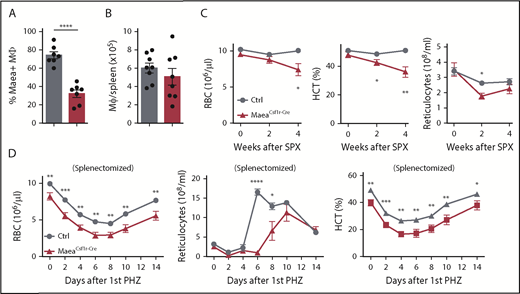
Maea deletion impairs BM erythropoiesis under steady state and stress conditions. (A) Deletion efficiency of Maea by Csf1r-Cre in spleen macrophages as determined by FACS (n = 7). (B) Quantification of spleen RPM numbers (n = 8). (C) RBC, hematocrit (HCT), and reticulocyte measurements of control and MaeaCsf1r-Cre mice over the course of 4 weeks after splenectomy (n = 9). (D) RBC, HCT, and reticulocyte assessment in splenectomized control and MaeaCsf1r-Cre mice after PHZ-induced hemolytic anemia (n = 9 per group, pooled from 2 independent experiments). Data are shown as mean plus or minus SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001 by unpaired Student t test. SPX, splenectomy.
Maea deletion impairs BM erythropoiesis under steady state and stress conditions. (A) Deletion efficiency of Maea by Csf1r-Cre in spleen macrophages as determined by FACS (n = 7). (B) Quantification of spleen RPM numbers (n = 8). (C) RBC, hematocrit (HCT), and reticulocyte measurements of control and MaeaCsf1r-Cre mice over the course of 4 weeks after splenectomy (n = 9). (D) RBC, HCT, and reticulocyte assessment in splenectomized control and MaeaCsf1r-Cre mice after PHZ-induced hemolytic anemia (n = 9 per group, pooled from 2 independent experiments). Data are shown as mean plus or minus SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001 by unpaired Student t test. SPX, splenectomy.
To further dissect the requirement of Maea in BM and spleen erythropoiesis, we splenectomized MaeaCsf1r-Cre and littermate control mice. Splenectomized MaeaCsf1r-Cre animals developed anemia over the course of 4 weeks whereas the control group maintained healthy peripheral blood counts (Figure 3C), suggesting that in the context of MaeaCsf1r-Cre animals, the extramedullary splenic erythropoiesis may mask the phenotype. We next challenged splenectomized control and MaeaCsf1r-Cre mice with PHZ and found a severely impaired recovery response in MaeaCsf1r-Cre mice (Figure 3D). Blood smear again did not reveal any enucleation defect in the RBCs from splenectomized MaeaCsf1r-Cre mice before or after PHZ treatment (data not shown). These results further confirm that Maea is critical for adult BM erythropoiesis but not RBC enucleation.
Dominant function of Maea over Vcam1 in EI niche formation
Vcam1 represents another adhesion molecule implicated in EI.8,9,19 To compare the role of Vcam1 with Maea, we also deleted Vcam1 using Csf1r-Cre. Interestingly, we did not observe defects in BM macrophage or EB numbers in steady-state Vcam1Csf1r-Cre mice compared with the control animals, except for a minor trend toward reduced spleen EBs (supplemental Figure 5A-C). EB maturation as measured by CD44 expression and cell size also indicated a normal differentiation profile (supplemental Figure 5D). The peripheral RBC compartment during steady state (supplemental Figure 5E) and after PHZ challenge (supplemental Figure 5F) was also normal, consistent with previous studies.29,30 These data indicate that Maea plays a dominant role in BM EI niche function whereas Vcam1 is dispensable for adult EI function in vivo.
Selective Maea deletion in macrophages, but not EBs, impairs BM erythropoiesis
We found that Csf1r-Cre broadly recombines in hematopoietic stem and progenitor cells (HSPCs; Figure 4A), and thus it may not discriminate between Maea function in macrophages or EBs that descend from HSPCs. To delete Maea in macrophages selectively, we crossed Maeafl/fl with CD169-Cre,17 which does not target the erythroid lineage (Figure 4B). MaeaCD169-Cre mice phenotypically mimicked the MaeaCsf1r-Cre animals, with significant reduction of BM macrophage and EB numbers, but no alterations in BM cellularity or circulating blood parameters (Figure 4C-D). Further analyses revealed a similar defect in in vivo island formation and a partial block in BM EB maturation at the EB-III stage (Figure 4F-G). Interestingly, although CD169-Cre also recombined efficiently in spleen RPMs (Figure 4H), no significant change in RPM numbers was observed and EB numbers in the spleen of MaeaCD169-Cre mice were significantly increased, suggesting ongoing extramedullary erythropoiesis (Figure 4I-K).
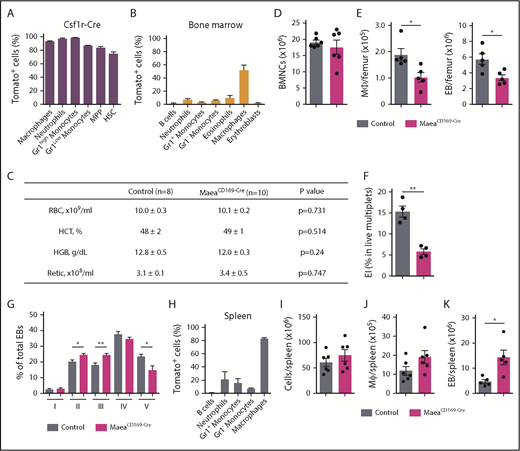
Deletion of Maea expressed by macrophages impairs BM EI niche. (A) Expression pattern and recombination efficiency of Csf1r-Cre in BM cells of R26TdTomato reporter mice (n = 3). (B) Expression pattern and recombination efficiency of CD169-Cre in BM cells of R26TdTomato reporter mice (n = 3). (C) RBC, hematocrit (HCT), hemoglobin (HGB), and reticulocyte (Retic) assessment of steady state control and MaeaCD169-Cre mice. (D) Total BM cellularity of control and MaeaCD169-Cre mice (n = 6). (E) Quantification of BM macrophages and EBs in MaeaCD169-Cre mice and littermate controls (n = 6). (F) Quantification of in vivo EIs (EI) in MaeaCD169-Cre and control BM (n = 4). (G) Quantification of EBs at various stages of maturation from control and MaeaCD169-Cre BM (n = 6). (H) Expression pattern and recombination efficiency of CD169-Cre in splenocytes of R26TdTomato reporter mice (n = 3). (I-K) Quantifications of total cellularity (I), macrophage (J), and EB (K) numbers in control and MaeaCD169-Cre spleens (n = 6). Data are shown as mean plus or minus SEM. *P < .05, **P < .01 by unpaired Student t test. HSC, hematopoietic stem cell; MPP, multipotent progenitors.
Deletion of Maea expressed by macrophages impairs BM EI niche. (A) Expression pattern and recombination efficiency of Csf1r-Cre in BM cells of R26TdTomato reporter mice (n = 3). (B) Expression pattern and recombination efficiency of CD169-Cre in BM cells of R26TdTomato reporter mice (n = 3). (C) RBC, hematocrit (HCT), hemoglobin (HGB), and reticulocyte (Retic) assessment of steady state control and MaeaCD169-Cre mice. (D) Total BM cellularity of control and MaeaCD169-Cre mice (n = 6). (E) Quantification of BM macrophages and EBs in MaeaCD169-Cre mice and littermate controls (n = 6). (F) Quantification of in vivo EIs (EI) in MaeaCD169-Cre and control BM (n = 4). (G) Quantification of EBs at various stages of maturation from control and MaeaCD169-Cre BM (n = 6). (H) Expression pattern and recombination efficiency of CD169-Cre in splenocytes of R26TdTomato reporter mice (n = 3). (I-K) Quantifications of total cellularity (I), macrophage (J), and EB (K) numbers in control and MaeaCD169-Cre spleens (n = 6). Data are shown as mean plus or minus SEM. *P < .05, **P < .01 by unpaired Student t test. HSC, hematopoietic stem cell; MPP, multipotent progenitors.
By contrast, when we crossed Maeafl/fl with Epor-Cre, which recombines efficiently and selectively in erythroid progenitors18 (Figure 5A; supplemental Figure 6A-B), MaeaEpor-Cre animals showed significant reductions in circulating RBC counts with increased mean corpuscular volume (Figure 5B; supplemental Figure 6C), but no significant change in BM macrophage and EB numbers (Figure 5C-D) or in vivo EB island formation (Figure 5E; supplemental Figure 6D). In addition, MaeaEpor-Cre EB maturation showed a distinct profile with accumulation of the mature cells in the BM (Figure 5F). No enucleation defect was observed in blood smear or in vitro EB-III culture (supplemental Figure 6E). Analysis of the spleen did not reveal significant differences in macrophage and EB numbers in MaeaEpor-Cre mice, despite efficient Epor-Cre recombination in spleen erythroid lineage (Figure 5G-J). These results suggest that Maea acts in the macrophage, but not EB, to mediate EI formation in adult BM, consistent with the previous report in an in vitro EI reconstitution setting.14 Our result also further indicates that Maea is dispensable for RBC enucleation, but may play a role in regulating RBCs’ terminal maturation in adult mice.
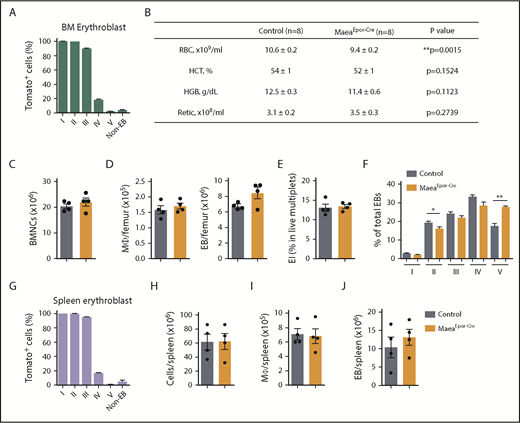
Maea expression by EBs is dispensable for BM EI niche. (A) Expression pattern and recombination efficiency of Epor-Cre in BM of R26TdTomato reporter mice (n = 3). (B) RBC, hematocrit (HCT), hemoglobin (HGB), and reticulocyte assessment of steady state control and MaeaEpor-Cre mice. (C) Total BM cellularity of control and MaeaEpor-Cre mice (n = 4). (D) Macrophage and EB numbers in control and MaeaEpor-Cre BM (n = 4). (E) Quantification of in vivo EI formation in MaeaEpor-Cre and control BM (n = 4). (F) Quantification of EBs at various stages of maturation from control and MaeaEpor-Cre BM (n = 4). (G) Expression pattern and recombination efficiency of Epor-Cre in spleen EBs and total splenocytes from R26TdTomato reporter mice (n = 3). (H-J) Quantifications of spleen total cellularity (H), spleen macrophage (I), and EB (J) numbers in control and MaeaEpor-Cre mice (n = 4). Data are shown as mean plus or minus SEM. *P < .05, **P < .01 by unpaired Student t test.
Maea expression by EBs is dispensable for BM EI niche. (A) Expression pattern and recombination efficiency of Epor-Cre in BM of R26TdTomato reporter mice (n = 3). (B) RBC, hematocrit (HCT), hemoglobin (HGB), and reticulocyte assessment of steady state control and MaeaEpor-Cre mice. (C) Total BM cellularity of control and MaeaEpor-Cre mice (n = 4). (D) Macrophage and EB numbers in control and MaeaEpor-Cre BM (n = 4). (E) Quantification of in vivo EI formation in MaeaEpor-Cre and control BM (n = 4). (F) Quantification of EBs at various stages of maturation from control and MaeaEpor-Cre BM (n = 4). (G) Expression pattern and recombination efficiency of Epor-Cre in spleen EBs and total splenocytes from R26TdTomato reporter mice (n = 3). (H-J) Quantifications of spleen total cellularity (H), spleen macrophage (I), and EB (J) numbers in control and MaeaEpor-Cre mice (n = 4). Data are shown as mean plus or minus SEM. *P < .05, **P < .01 by unpaired Student t test.
To investigate the role of macrophage or EB conditional Maea deletion in stress erythropoiesis, we challenged these 2 models with PHZ-induced anemia. Surprisingly, MaeaCD169-Cre mice showed no significant difference in hematocrit recovery or reticulocytosis (supplemental Figure 7A). This may be due to the fact that CD169-Cre recombines at a lower frequency (∼60%) in BM macrophages (Figure 4B), and/or the Csf1r-Cre model is indeed a combinatory model of Maea deletion in both the macrophages and EBs. Indeed, BM of MaeaCD169-Cre mice showed consistently milder reductions of macrophage and EB numbers than MaeaCsf1r-Cre mice at steady state (Figure 4E) and after PHZ (supplemental Figure 7C). In contrast, MaeaEpor-Cre mice showed a faster decline in RBC content during the early days and a weaker reticulocyte output during the later recovery stages (supplemental Figure 7B). The reduction of RBCs and reticulocytes but normal BM EB numbers after PHZ (supplemental Figure 7D) in MaeaEpor-Cre mice further suggests that EB Maea expression may regulate RBC terminal maturation or release from the EI.
Postnatal Maea deletion or inhibition uncovers important functions in adult erythropoiesis
To evaluate the role of Maea in postnatal erythropoiesis and the maintenance of the EI niche, we deleted Maea using the Mx1-Cre line (MaeaMx1-Cre) in which Cre-mediated recombination is inducible by Poly I:C injections. We firstly generated radiation chimeras by transplantation of the BM from MaeaMx1-Cre or littermate controls into wild-type mice (Figure 6A) to exclude a potential contribution from the BM microenvironment. Two months after transplantation, we induced Cre recombination by 3 Poly I:C injections. Three weeks after the first Poly I:C injection, BM macrophage and EB numbers were significantly reduced in MaeaMx1-Cre animals (Figure 6B), indicating that Maea was required for the postnatal maintenance of BM macrophages and the EI niche during homeostasis.
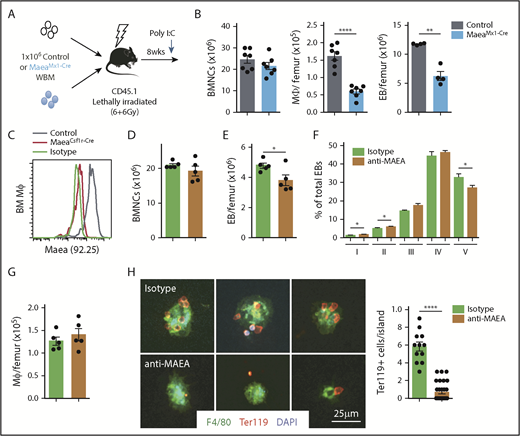
Maea maintains the postnatal EI. (A) Experimental design to determine the requirement of Maea during adult erythropoiesis using MaeaMx1-Cre mice. (B) Quantification of total BMNCs, macrophages, and EBs in Maeafl/fl and MaeaMx1-Cre mice 3 weeks after the first Poly I:C injection (n = 7; data pooled from 2 independent experiments). (C) Validation of the specificity of mAb produced by hybridoma clone 92.25 by FACS staining of BM macrophages from wild-type control and MaeaCsf1r-Cre mice. (D-G) Quantification of total BM cellularity (D), BM EB numbers (E), EB maturation profile (F), and BM macrophage numbers (G) in isotype or anti-MAEA mAb-treated mice (n = 5). (H) Representative immunofluorescence images and quantification of erythroid cells per EI reconstituted in the presence of 10 μg/mL isotype or anti-MAEA mAb. Data are shown as mean plus or minus SEM. *P < .05, **P < .01, ****P < .0001 by unpaired Student t test. WBM, whole bone marrow.
Maea maintains the postnatal EI. (A) Experimental design to determine the requirement of Maea during adult erythropoiesis using MaeaMx1-Cre mice. (B) Quantification of total BMNCs, macrophages, and EBs in Maeafl/fl and MaeaMx1-Cre mice 3 weeks after the first Poly I:C injection (n = 7; data pooled from 2 independent experiments). (C) Validation of the specificity of mAb produced by hybridoma clone 92.25 by FACS staining of BM macrophages from wild-type control and MaeaCsf1r-Cre mice. (D-G) Quantification of total BM cellularity (D), BM EB numbers (E), EB maturation profile (F), and BM macrophage numbers (G) in isotype or anti-MAEA mAb-treated mice (n = 5). (H) Representative immunofluorescence images and quantification of erythroid cells per EI reconstituted in the presence of 10 μg/mL isotype or anti-MAEA mAb. Data are shown as mean plus or minus SEM. *P < .05, **P < .01, ****P < .0001 by unpaired Student t test. WBM, whole bone marrow.
To investigate further the role of Maea in erythropoiesis, we developed a novel monoclonal antibody by immunization of Balb/c mice with a peptide corresponding to the extracellular domain of human MAEA. We isolated clone 92.25 (IgG2a), which interacts specifically with both murine and human MAEA, owing to the highly conserved MAEA amino acid sequence across species (Figure 6C). We then treated wild-type mice with 92.25 or isotype control (100 μg daily 5 days a week for 3 weeks) and found that anti-MAEA significantly reduced EB numbers in BM without affecting the total cellularity (Figure 6D-E), but not in the spleen (data not shown). The treatment also led to alterations in EB differentiation similar to macrophage-selective Maea knockouts (Figure 6F), without reductions of macrophage numbers (Figure 6G). Furthermore, an in vitro EI reconstitution assay showed that 92.25 significantly inhibited island formation (Figure 6H), clearly indicating that the EI phenotype from Maea deficiency does not originate solely from a defect of macrophage maturation and that direct adhesion mediated via Maea may play a role in adult BM erythropoiesis.
Discussion
The critical function of the EI niche in erythropoiesis was initially suggested based on in vitro data6,7 and was recently confirmed in vivo.19,31 However, in vivo studies using macrophage depletion cannot distinguish between the EI-dependent and EI-independent functions of the macrophage.19,31 Several adhesion mechanisms have been proposed to mediate EI formation, providing an ideal model for investigations of EI-specific functions.7,8,10-12 However, studies thus far have been largely based on in vitro EI formation assays or germline gene deletions. Here, we show that Maea is critical for adult BM EI formation and homeostatic erythropoiesis via multiple mechanisms. Both constitutive and induced Maea deletion led to severe reductions of BM macrophage numbers, indicating that Maea is required for BM macrophage homeostasis. Interestingly, spleen macrophages are not affected by Maea deletion, in line with recent reports indicating the independence and heterogeneity of tissue-resident macrophages under steady state.32-34 Accordingly, spleen EB numbers are unaltered but their differentiation is partially blocked in Maea-deleted mice, further suggesting that different mechanisms may regulate the spleen erythropoiesis. Indeed, studies have shown that splenic stress erythropoiesis uses a distinct erythroid progenitor population that responds to different signals than steady-state erythropoiesis.35 Although macrophages are required for spleen erythropoiesis,19 their exact identity and functions remain largely undefined.29,36 Yet, RBC clearance is delayed in MaeaCsf1r-Cre mice, suggesting a role of macrophages in the BM or other organs that may be dependent on Maea for RBC clearance. Additionally, the disruption of EI formation in vivo and in vitro by antibody inhibition also supports a previously suggested adhesion function between the EB and the macrophage to form EI.13,14 BM EB number and maturation profile are significantly impaired even when macrophage numbers are not affected, suggesting EI-specific functions of the macrophage in supporting EB differentiation.
Our macrophage and erythroid lineage-selective Maea deletion provides genetic evidence that Maea-mediated adhesion is unlikely the result of a homophilic interaction. The binding partner of Maea on erythroid progenitors is therefore yet to be characterized. The extracellular domain of Maea protein is highly conserved among vertebrates but does not contain discernable domains to identify its protein family. More rigorous proteomic studies will be needed to understand the adhesion mechanism of Maea.
Maea appears dispensable for the enucleation of adult EBs. The enucleation process of end-stage erythroid maturation is thought to be coordinated by the sorting and reassembly of nuclear, cytoplasmic, and membrane contents among the resulting reticulocytes and pyrenocytes.22,37,38 Previous studies have suggested that Maea is associated with actin filaments, preferentially segregating into the extruding pyrenocytes and is required for EB enucleation.14,38,39 However, none of our genetic deletion models have revealed any enucleation defect in vivo or in vitro, under steady state or after stress. One possibility is that the erythroid progenitors in previously reported Maea-null embryos are so poorly differentiated, due to defects upstream of the EI, that they are not able to reach the enucleation stage.14,39 Alternatively, the residual expression of Maea in our conditional knockout models may be masking the phenotype observed in the null embryos. It is also tempting to speculate that the enucleation process of fetal erythrocytes may be different from their adult counterpart, like with many other differences, such as the globin composition.40,41
Our current and previous results19 also suggest that the EI niche may not be the only erythroid niche because macrophage or Maea-depleted mice are still able to respond to erythroid stress. Additional niche cells (eg, EPO-secreting Osx+ osteolineage42 or Vcam1+43 stromal cells, and SCF-producing type 1 conventional dendritic cells44 ) and secreted factors (eg, SCF, BMP4, and EPO)35,45 have indeed been reported to regulate erythropoiesis. We have recently identified a novel population of CD45−Ter119− erythroid progenitors whose survival and expansion are dependent on BM-derived stromal cells.46 It is intriguing to hypothesize that different erythroid niches may exist to support distinct erythroid progenitor subsets or differentiation pathways.
Our studies also provide genetic evidence that the contribution of Maea in BM EI is dominant compared with that of Vcam1, which, when deleted using the same Csf1r-Cre, is dispensable for macrophage development, in vivo EI function, and erythroid recovery. Although Vcam1-mediated EI formation has commonly been observed in vitro,8,47,48 its requirement during in vivo erythropoiesis using genetic models has not been described.29,30 Studies using antibody inhibition in whole animals have suggested a contribution of Vcam1 in erythropoiesis,19 although Vcam1 expression and function outside of the EI, for example, in endothelial cells49 or the HSPC niche,43,50,51 could not be excluded in this systemic inhibition model. Because EI mediates erythropoiesis in health and disease,19,31 our study indicates that MAEA may be a promising therapeutic target for erythropoietic disorders.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Y. Zhang of the Albert Einstein College of Medicine Gene Targeting and Transgenic Core, S. V. Buhl and Scharff of the Macromolecule Therapeutics Core, and L. Tesfa of the Flow Cytometry Sorting Facility for technical assistance and guidance.
This work was supported by the National Institutes of Health (DK056638 [National Institute of Diabetes and Digestive and Kidney Diseases]; HL069438 and HL116340 [National Heart, Lung, and Blood Institute]), the Leukemia & Lymphoma Society (LLS-TRP 6475-15), and the New York State Department of Health (NYSTEM IIRP C029570 and C029154).
Authorship
Contribution: Q.W. performed most of the experiments, analyzed data, and wrote the manuscript; P.E.B. helped analyze the MaeaCD169-Cre mice; D.Z. performed splenectomy of the MaeaCsf1r-Cre mice; S.P. helped analyze the Vcam1Csf1r-Cre mice; M.T. contributed the CD169-Cre mice; P.S.F. supervised the study and wrote the manuscript; and all authors discussed the results and contributed to the manuscript.
Conflict-of-interest disclosure: Q.W. and P.S.F. are coinventors on a patent application using anti-MAEA antibodies (W02017205560A1). P.S.F. serves as a consultant for Pfizer, has received research funding from Ironwood Pharmaceuticals, and is a shareholder of Cygnal Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Paul S. Frenette, Albert Einstein College of Medicine, Price Center, Room 101B, 1301 Morris Park Ave, New York, NY 10461; e-mail: paul.frenette@einstein.yu.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal