

Abstract
Acute myeloid leukemia (AML) is the most frequent and heterogeneous malignancy in adult leukemic patients. Genome-wide analyses revealed that genes involved in epigenetic modifications are among the most often re-occurring mutations in AML, suggesting a crucial role of epigenetic regulation in leukemogenesis and leukemia relapse. As a mammalian lysine methyltransferase, SUV39H1 catalyzes di- and tri-methylation of histone 3 lysine 9, and is the predominant H3K9 methyltransferase expressed in hematopoietic stem cells (HSCs). Previous studies have shown that in MLL-rearranged leukemic cells, the normal localization of Suv39h1 and Sirt1 was interrupted due to the DNA binding of Dot1L to DNA. However, the biological role of SUV39H1 in MLL-rearranged leukemia remains unexplored.
In this study, we investigated the role and the underlying mechanism of Suv39h1 during leukemia progression. By analyzing the clinical databases, we found a significantly reduced expression of SUV39H1 in AML cells in comparison with normal bone marrow (BM) cells. More importantly, we found that low expression of SUV39H1 predicts poorer survival in AML patients. In MLL-fusion induced AML mouse models (MLL-AF9/MA9 and MLL-NRIP3/MN3), Suv39h1 also exhibited lower expression in leukemia stem cells (LSCs, defined as c-Kit+ or Lin-Sca1-IL-7R-c-Kit+CD34+CD16/32+ L-GMP cells) when compared with normal HSPCs. These data suggest a potential role of SUV39H1 in leukemic progression and/or maintenance.
To explore if Suv39h1 functions as a tumor suppressor in MLL-fusion driven leukemogenesis, we overexpressed Suv39h1 in MA9 BM AML cells. Western blotting analysis confirmed the overexpression of Suv39h1 with a moderate increase in global H3K9me3 levels in Suv39h1-overexpressed (SUV-OE) MA9 AML cells. Interestingly, Suv39h1 overexpression prolonged the survival of recipient AML mice in both secondary and tertiary transplantation groups. Both the frequency and the absolute number of phenotypic LSCs in BM and SP were significantly reduced in SUV-OE groups as manifested by flow cytometry. Furthermore, limiting dilution assays revealed a significant six-fold decrease of functional LSCs in SUV-OE AML cells (1/314 LSCs in SUV-OE AML cells vs 1/56 in controls). Cell cycle analysis of control and SUV-OE LSCs from BM revealed a significantly decreased proportion of SUV-OE cells in the S/G2/M phase concordant by an increased proportion of G0/G1 phases when compared with control cells. In contrast, a similar apoptotic ratio of L-GMPs in BM was observed between control and SUV-OE groups. Taken together, these data demonstrated that overexpressing Suv39h1 in AML cells reduces the frequency of functional LSCs by suppression its proliferation.
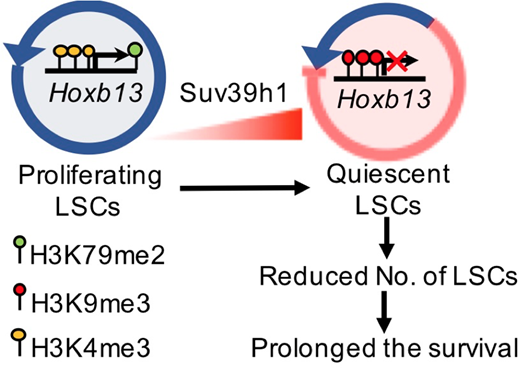
To explore the underlying mechanisms, gene expression profiles were assessed by RNA-Seq of SUV-OE and control mouse AML c-Kit+ cells. A total of 69 genes were differentially expressed with fold change ≥ 4. Among these genes, Hoxb13 was of particular interesting since it was reported to be recurrently mutated in several types of cancers including leukemia. ChIP-qPCR revealed a two-fold increase of H3K9m3 distribution at the promoter of Hoxb13 in SUV-OE groups, indicating Hoxb13 may be a direct downstream target of Suv39h1. Restoring the expression of Hoxb13 in SUV-OE AML cells diminished the effect of SUV-OE-mediated prolonged survival of SUV-OE AML mice. Interestingly, overexpression of Hoxb13 alone in MA9 cells had no significant effect on the survival of MA9 AML mice, indicating that Hoxb13 is a downstream effector of Suv39h1, rather than MA9, and Suv39h1 itself is a downstream mediator of MA9.
To summarize, we here for the first time, demonstrate that Suv39h1 is significantly down-regulated in AMLs and could function as a tumor suppressor in MLL-rearranged leukemia by epigenetically inhibiting the Hoxb13 expression. The molecular mechanism mediated by Suv39h1-Hoxb13 axis in tumor suppression could potentially provide us novel therapeutic strategies for MLL-rearranged leukemia.
YJ.C, YP.C and HD.G contributed equally to this work.
Corresponding authors: WP.Y and MJ.X.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal