

Key Points
Direct Rap1/Talin1 interaction controls platelet and neutrophil integrin functions.
Rap1/Talin1 interaction is important in blood cells, which depend on rapid integrin-mediated processes.

Abstract
Targeting Talin1 to the plasma membrane is a crucial step in integrin activation, which in leukocytes is mediated by a Rap1/RIAM/Talin1 pathway, whereas in platelets, it is RIAM independent. Recent structural, biochemical, and cell biological studies have suggested direct Rap1/Talin1 interaction as an alternative mechanism to recruit Talin1 to the membrane and induce integrin activation. To test whether this pathway is of relevance in vivo, we generated Rap1 binding–deficient Talin1 knockin (Tln13mut) mice. Although Tln13mut mice showed no obvious abnormalities, their platelets exhibited reduced integrin activation, aggregation, adhesion, and spreading, resulting in prolonged tail-bleeding times and delayed thrombus formation and vessel occlusion in vivo. Surprisingly, neutrophil adhesion to different integrin ligands and β2 integrin–dependent phagocytosis were also significantly impaired, which caused profound leukocyte adhesion and extravasation defects in Tln13mut mice. In contrast, macrophages exhibited no defect in adhesion or spreading despite reduced integrin activation. Taken together, our findings suggest that direct Rap1/Talin1 interaction is of particular importance in regulating the activity of different integrin classes expressed on platelets and neutrophils, which both depend on fast and dynamic integrin-mediated responses.
Introduction
Integrins can adopt different conformations with different affinities for their ligands. Blood cell integrins become activated upon direct binding of the 2 adapter proteins Talin1 and Kindlin-3 to the cytoplasmic domain of integrin β subunit. Rapid regulation of integrin activity is particularly important for integrins expressed on platelets, which control thrombosis and hemostasis at sites of vascular injury, and for leukocyte integrins, which are essential for leukocyte adhesion and extravasation at sites of inflammation. A critical but as yet incompletely understood process during integrin activation is the recruitment of these activators from the cytoplasm to the plasma membrane. A series of in vitro studies on the prototypic integrin αIIbβ3 from platelets suggested that membrane-bound, guanosine triphosphate (GTP)–loaded Rap1 binds its effector protein Rap1 GTP-interacting adapter molecule (RIAM), which in turn binds Talin1 and thereby recruits it to the plasma membrane to activate integrins.1-4 However, although leukocyte β2 integrin activation is dependent on the Rap1/RIAM/Talin1 pathway, platelet integrins become normally activated in RIAM knockout mice, suggesting the existence of a RIAM-independent Talin1 recruitment pathway.5-10 On the basis of previous studies, which showed that the F0 domain of Talin1 can interact with Rap1 weakly and is required for maximal integrin activation,11-13 we recently provided strong evidence that the direct Rap1/Talin1 interaction becomes stabilized when Rap1 is membrane anchored and that the disruption of such an interaction diminishes Talin1 recruitment to the plasma membrane, resulting in reduced integrin activation and signaling in vitro.14 Here we report a thorough analysis of Rap1 binding–deficient Talin1 knockin mice and demonstrate the importance of such interactions for both platelet and neutrophil functions in vivo.
Materials and methods
Mice
Tln13mut mice were generated by injection of correctly targeted R1 embryonic stem cells into C57BL/6 blastocysts. The resulting chimeric mice were crossed with deleter-Cre mice to remove the loxP-flanked neomycin cassette.15 Mouse experiments were performed with approval of the District Government of Bavaria.
Platelet in vitro assays
Integrin activation and platelet aggregation,16 platelet spreading,17 and platelet filamentous actin content measurement18 were in essence carried out as previously described. Platelet adhesion under flow was measured using fibrillar collagen–coated (10 µg/mL; Horm, Takeda Austria GmbH, Linz, Austria) Ibidi µ-Slides VI 0.1, uncoated (Ibidi, Martinsried, Germany). Chambers were perfused with heparinized blood for 4 minutes and subsequently washed with Tyrode’s solution at a shear rate of 1000/s. Images were taken with a Zeiss Axio Imager Z1 microscope equipped with a 20× NA 0.75 objective and an AxioCam MRm (Carl Zeiss, Inc., Oberkochen, Germany) after staining with a CellTrace CFSE Cell Proliferation Kit (Thermo Fisher Scientific). Pictures were processed and surface coverage was measured using ImageJ software (US National Institutes of Health) and normalized against platelet counts.
Microvascular thrombosis and tail bleeding
The surgical preparation of the mouse cremaster muscle was performed as described previously by Baez19 with minor modifications. Mice were anesthetized by intraperitoneal administration of a mixture of 100 mg/kg of ketamine and 10 mg/kg of xylazin. The left femoral artery was cannulated in a retrograde manner for the administration of fluorescein isothiocyanate–labeled dextran (molecular weight, 150 kDa; Sigma Aldrich) as well as of DyLight 649-labeled antibodies directed against GPIbβ for the visualization of platelets (EMFRET, Eibelstadt, Germany). The right cremaster muscle was exposed through a ventral incision of the scrotum, then opened and spread over the pedestal of a custom-made microscopy stage. Epididymis and testicle were detached from the cremaster muscle and placed back into the abdominal cavity. Throughout the surgical procedure and during in vivo microscopy measurements, the cremaster muscle was continuously perfused with warm buffered saline. Microvascular thrombus formation was induced by photochemical injury as described previously.20 Briefly, a 2.5% solution of fluorescein isothiocyanate–dextran was infused intraarterially, and the exposed vessel segment under investigation was continuously epi-illuminated with a wavelength of 488 nm. An Axio Scope.A1 microscope (Carl Zeiss Microscopy GmbH, Goettingen, Germany), equipped with a Colibiri 2 LED light source (Carl Zeiss Microscopy GmbH) for fluorescence epi-illumination microscopy, was used to visualize microvascular thrombogenesis. Thrombus formation was induced in 3 venules and 1 arteriole (inner diameter, 20-30 μm) per experiment. Time until the first platelet adhesion to the vessel wall (defined as the onset of thrombus formation) and time until blood flow ceased (defined as the complete occlusion of the vessel) were analyzed.
To study thrombus formation in larger vessels, the tails of anesthetized mice were transected at a 5-mm distance from the tail tip using a sterile surgical scalpel blade. Subsequently, the tail was immersed in warm buffered saline (37°C), and tail bleeding was monitored until bleeding ceased. The end point was the arrest of bleeding lasting >6 seconds. Bleeding was recorded for a maximum of 15 minutes.
Leukocyte in vitro assays
Neutrophils were isolated from bone marrow using an EasySep Mouse Neutrophil Enrichment Kit (STEMCELL Technologies) following the manufacturer’s instructions.
Bone marrow–derived macrophages for in vitro assays were differentiated by culturing bone marrow cells in RPMI 1640 supplemented with 10% fetal bovine serum, 100 U/mL of penicillin, 100 µg/mL of streptomycin (all Thermo Fisher Scientific), 50 µM of β-mercaptoethanol, and macrophage colony-stimulating factor for 6 days. Neutrophil and macrophage adhesion and spreading assays were performed as previously described.10
Polymorphonuclear leukocyte (PMN) adhesion strengthening was analyzed using ibidi µ-Slides VI 0.4, uncoated (Ibidi), coated with 12.5 µg/mL of recombinant human intercellular cell adhesion molecule 1 (ICAM-1; R&D Systems). A neutrophil suspenstion of 3 × 106 cells per mL isolated from bone marrow using an EasySep Mouse Neutrophil Enrichment Kit (STEMCELL Technologies) was stained using a CellTrace CFSE Cell Proliferation Kit (Thermo Fisher Scientific). PMNs were stimulated with 2 µg/mL of tumor necrosis factor α (TNF-α; R&D Systems), placed into flow chambers, and allowed to adhere for 10 minutes. Subsequently, shear stress of 0.5, 1, 2, 4, 6, and 9 dyn/cm2 was applied, gradually increasing every 90 seconds, and time-lapse movies were recorded using an Evos FL Auto 2 life cell microscope (Thermo Fisher Scientific) equipped with an Evos FL 10× NA 0.30 objective. The number of adherent cells was counted and normalized to the number of initially adherent cells for all applied shear rates using Photoshop CS6 (Adobe).
Intravital microscopy of TNF-α–stimulated mouse cremaster muscle venules
Mice were treated by intrascrotal injection of 500 ng of TNF-α (R&D Systems) 2 hours before microscopy, anesthetized, and prepared for intravital microscopy as described.21 Movies from cremasteric postcapillary venules ranging from 20 to 40 μm in diameter were recorded using a BX51WI microscope with a water immersion objective of 40× 0.80 NA and an Olympus CCD camera (CF8/1, κ). Blood samples were taken after the experiment. White blood cell and neutrophil counts were determined using ProCyte Dx Hematology Analyzer (IDEXX, Westbrook, ME). Leukocyte rolling flux fraction (percentage of rolling leukocytes in all leukocytes passing through the vessel), rolling velocity, adhesion, and adhesion efficiency (number of adherent cells per mm2 divided by the systemic neutrophil count) were calculated based on the recorded movies using Fiji software.22 Afterward, cremaster muscles were fixed with 4% paraformaldehyde (AppliChem GmbH, Darmstadt, Germany) and stained using Giemsa (Merck Millipore, Darmstadt, Germany). The number of perivascular cells per mm2 was assessed with a Leica DM2500 microscope equipped with a DMC2900 CMOS camera and an HCX PL APO 100×/1.40 Oil Ph3 at the Ludwig Maximilians University Munich Biomedical Center BioImaging core facility.
Statistical analysis
Data are presented as means ± 95% confidence intervals or standard errors of the mean. Paired or unpaired Student’s t tests were used to compare data sets. A difference between data sets was considered to be significant if P < .05.
Additional methods and more detailed descriptions are provided in the supplemental Material and methods, available on the Blood Web site.
Results
Rap1-binding mutant Talin1 knockin mice are viable and appear healthy
To interfere with Rap1/Talin1 interaction in vivo, we generated a Talin1 knockin mouse strain (Tln13mut) carrying mutations within exon 2, where K15, R30, and R35 in the Talin1-F0 domain were changed into alanines (supplemental Figure 1A). We chose to mutate these 3 basic residues based on the previous surface charge analysis that suggested their involvement in binding to Rap1.12 While we generated Tln13mut knockin mice, the complex structure of Rap1/Talin1-F0 was determined, which revealed that K15 and R35 are indeed part of the binding interface and their mutations into alanines (Talin1-F0_DM) are sufficient to block Rap1 binding.14 R30 is located near the binding interface; although not directly involved in Rap1 binding, its mutation into alanine should exert little effect on the folding of Talin1-F0.14 By NMR-based experiments, we show that Talin1-F0 bearing K15A/R30A/R35A (Talin1-F0_3mut) exhibit a similar NMR spectrum to that of wild-type (WT) Talin1-F0 domain (Talin1-F0_WT) and the previously reported K15A/R35A (Talin1-F0_DM) mutant14 demonstrating no perturbation in the overall folding of mutant Talin1-F0 domains (supplemental Figure 2A-B). As expected, the Talin1-F0_3mut, like the Talin1-F0_DM, did not bind to Rap1b (supplemental Figure 2C). This is consistent with the cellular data, which show that Talin1 and Talin2 double-deficient (Talin1/2dko) fibroblasts with retroviral expression of full-length ypet-tagged Talin1_3mut or Talin1_DM exhibited identical defects in cell adhesion and spreading compared with those with Talin1_WT (supplemental Figure 2D-E). Importantly, similar to Talin1_DM, the Talin1_3mut also showed significantly reduced recruitment to focal adhesions (FAs) (supplemental Figure 2F).
Lagarrigue et al23 recently reported single point mutant Talin1 knockin (R35E) mice, which exhibited a significantly milder phenotype than ours. It remains unclear what caused the discrepancy, but a strong possibility is the weaker disruption of Rap1/Talin1 interaction by the single R35E mutation compared with our Talin1-F0_3mut. This explanation is supported by the overall chemical shift changes of 15N-labeled Talin1-F0 variants, including WT, K15A, R35A, R35E, DM, and 3mut, in the presence of GMP-PNP–loaded Rap1b, where single mutants, including R35E, of Talin1-F0 indeed showed much less reduced binding to Rap1 than Talin1-F0_DM or 3mut (supplemental Figures 3A-D and 4A-F). That is, the residual interaction between Rap1 and Talin1 (R35E) may still play an important role in integrin signaling, thereby leading to the milder phenotype observed in Talin1 (R35E) knockin mice. Our Tln13mut mice are clearly better suited to explore the physiological relevance of Rap1/Talin1 interaction in regulating integrin activity, even though a residual interaction between Rap1 and Talin1_3mut may still exist in the cellular context, because we found that expression of Talin1 lacking the entire F0 domain led to more dramatic adhesion, spreading, and recruitment defects in Talin1/2dko fibroblasts than the DM or 3mut mutant (supplemental Figure 2D-F).
Heterozygous Tln13mut mice were intercrossed to obtain homozygous Tln13mut mice, and their mutations were confirmed by genomic polymerase chain reaction followed by sequencing (supplemental Figure 1B). Tln13mut mice were born at a normal Mendelian ratio (supplemental Table 1), were viable, and did not show any overt phenotype. The lack of any obvious phenotype was indeed surprising, given that our previous study revealed a robust integrin-signaling defect in Talin1/2dko fibroblasts expressing a Rap1 binding–deficient Talin1.14 Our reasoning was that Talin2 might compensate for the integrin defect in Tln13mut mice. Indeed, we found that mouse embryonic fibroblasts (MEFs) and endothelial cells isolated from WT and Tln13mut mice expressed Talin2, which was not detectable in blood cells (supplemental Figure 5A). Consistently, MEFs from Tln13mut mice showed no difference in adhesion to the integrin ligands fibronectin and vitronectin or in the number or size of FAs (supplemental Figure 5B-F) as compared with MEFs isolated from WT embryos. Our hypothesis was further supported by the localization of Talin2 to FAs in both WT and mutant MEFs (supplemental Figure 5G).
We then focused our study on blood cells, which exhibit negligible Talin2 expression (supplemental Figure 5F). To exclude the possibility that targeting the Talin1 gene locus might alter Talin1 expression, we carefully measured Talin1 protein levels in platelets and macrophages from WT and Tln13mut mice by western blotting and found them to be identical (supplemental Figure 6). Furthermore, platelet and white blood cell counts were indistinguishable between control and Tln13mut mice (supplemental Table 2). Altogether, these data suggest that Tln13mut is normally expressed in vivo, expression of Talin2 in nonhematopoietic cells might compensate for the defective Rap1/Talin1 interaction, and direct Rap1/Talin1 interaction is not essential for hematopoiesis.
Impaired integrin activity and signaling in Tln13mut platelets
Because Talin1 and Rap1 are highly expressed in platelets, whereas RIAM expression is much lower,24 we hypothesized that direct Rap1/Talin1 interaction is of particular importance for platelet integrin activity. We first determined the expression of Kindlin-3, RIAM, and Rap1 and the surface levels of integrins and found them comparable between platelets from WT and Tln13mut mice (Figure 1A-B). Next, we measured agonist-induced αIIbβ3 integrin activation using the conformation-specific JON/A-PE antibody.25 We found that stimulation of WT and Tln13mut platelets by high concentrations of thrombin or other agonists, such as ADP, U46619, and collagen-related peptide, results in a similar response. However, lower concentrations of thrombin or other agonists induced the activation of fewer integrins in Tln13mut platelets compared with WT controls, suggesting a reduced susceptibility of Tln13mut platelets in response to agonist stimulation (Figure 1C). Similar results were obtained in binding assays with Alexa Fluor 647–conjugated fibrinogen (Figure 1D). Consistently, Tln13mut platelets revealed similar aggregation responses compared with WT platelets when activated by high concentrations of agonists but showed defective aggregation upon stimulation with agonists at lower concentrations (Figure 1E; supplemental Figure 7). To test whether α2β1 integrin activation is also diminished in Tln13mut platelets, we performed platelet adhesion assays on collagen under flow and observed significantly reduced adhesion (Figure 1F-G). We also allowed control and Tln13mut platelets to adhere and spread on fibrinogen and analyzed the spreading kinetics by measuring the spreading area over time and found delayed spreading in Tln13mut platelets (Figure 1H). A more detailed characterization revealed that the spreading defect is caused by delayed filopodia and lamellipodia formation resulting from reduced actin polymerization (Figure 1I-J), suggesting that integrin outside-in signaling is also significantly impaired in Tln13mut platelets. Collectively, our data demonstrate an important role of direct Rap1/Talin1 interaction in regulating platelet aggregation, adhesion, and spreading.
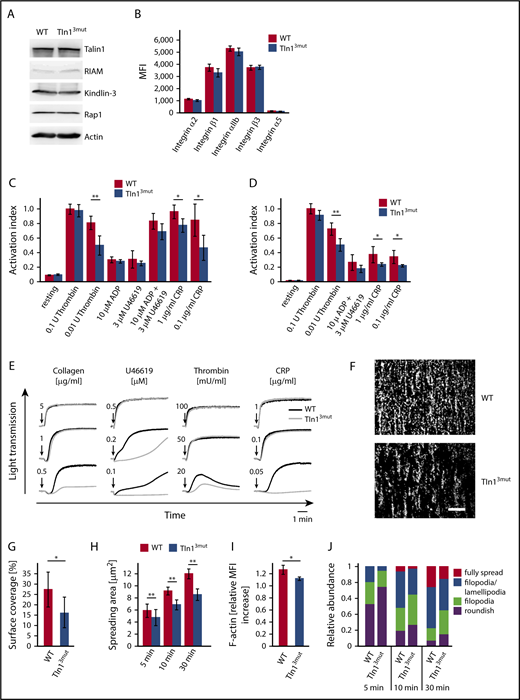
Platelet function is impaired in Tln13mutmice. (A) Western blots showing Talin1, RIAM, Kindlin-3, and Rap1 levels in WT and Tln13mut platelets. Actin served as loading control. (B) Surface levels of integrins α2, β1, αIIb, β3, and α5 assessed by flow cytometry (n = 4 mice). (C-D) Platelet integrin activation of control and Tln3mut platelets analyzed by JON/A (C) and fibrinogen binding (D) upon stimulation with thrombin (n = 11 WT/12 Tln13mut), ADP (n = 11 WT/12 Tln13mut), U46619 (n = 5), a combination of ADP and U46619 (n = 5), or collagen-related peptide (CRP; n = 5). Activation index represents mean fluorescence intensity (MFI) values of JON/A-PE or fibrinogen–Alexa 647 normalized to integrin β3 levels. MFI values of WT platelets stimulated with 0.1 U of thrombin are set to 1; n represents the number of mice. (E) Representative platelet aggregation curves measured upon stimulation with different concentrations of collagen, U46619, thrombin, and CRP. Quantification of the data is provided in supplemental Figure 5. (F-G) Heparinized whole blood was perfused through micro flow chambers coated with fibrillar collagen to assess adhesion under flow. (F) Representative images of adherent CFSE-stained platelets after perfusion. Scale bar, 80 μm. (G) Quantification of platelet surface coverage normalized to platelet counts (n = 7 mice). (H) Quantification of platelet spreading area on fibrinogen 5, 10, and 30 minutes after activation with 0.01 U of thrombin (n = 5 experiments and mice). (I) Relative F-actin levels in resting vs thrombin-activated (0.01 U of thrombin) platelets quantified by phalloidin–Alexa Fluor 546 staining and subsequent fluorescence-activated cell sorter analysis (n = 4 mice). (J) Relative distribution of WT and Tln13mut platelets at different spreading stages 5, 10, and 30 minutes after plating on a fibrinogen-coated surface (n = 5 experiments and mice). Values are given as means ± 95% confidence intervals. *P < .05, **P < .01.
Platelet function is impaired in Tln13mutmice. (A) Western blots showing Talin1, RIAM, Kindlin-3, and Rap1 levels in WT and Tln13mut platelets. Actin served as loading control. (B) Surface levels of integrins α2, β1, αIIb, β3, and α5 assessed by flow cytometry (n = 4 mice). (C-D) Platelet integrin activation of control and Tln3mut platelets analyzed by JON/A (C) and fibrinogen binding (D) upon stimulation with thrombin (n = 11 WT/12 Tln13mut), ADP (n = 11 WT/12 Tln13mut), U46619 (n = 5), a combination of ADP and U46619 (n = 5), or collagen-related peptide (CRP; n = 5). Activation index represents mean fluorescence intensity (MFI) values of JON/A-PE or fibrinogen–Alexa 647 normalized to integrin β3 levels. MFI values of WT platelets stimulated with 0.1 U of thrombin are set to 1; n represents the number of mice. (E) Representative platelet aggregation curves measured upon stimulation with different concentrations of collagen, U46619, thrombin, and CRP. Quantification of the data is provided in supplemental Figure 5. (F-G) Heparinized whole blood was perfused through micro flow chambers coated with fibrillar collagen to assess adhesion under flow. (F) Representative images of adherent CFSE-stained platelets after perfusion. Scale bar, 80 μm. (G) Quantification of platelet surface coverage normalized to platelet counts (n = 7 mice). (H) Quantification of platelet spreading area on fibrinogen 5, 10, and 30 minutes after activation with 0.01 U of thrombin (n = 5 experiments and mice). (I) Relative F-actin levels in resting vs thrombin-activated (0.01 U of thrombin) platelets quantified by phalloidin–Alexa Fluor 546 staining and subsequent fluorescence-activated cell sorter analysis (n = 4 mice). (J) Relative distribution of WT and Tln13mut platelets at different spreading stages 5, 10, and 30 minutes after plating on a fibrinogen-coated surface (n = 5 experiments and mice). Values are given as means ± 95% confidence intervals. *P < .05, **P < .01.
Rap1/Talin1 interaction is important for hemostasis and thrombosis
To investigate whether the loss of Rap1/Talin1 interaction in platelets is of any relevance in vivo, we first tested the hemostatic potential in Tln13mut mice by performing tail-bleeding assays, which revealed significantly prolonged bleeding time in Tln13mut mice compared with WT mice (Figure 2A). We also studied pathological thrombus formation at a phototoxic-induced vessel injury site in the cremaster muscle vasculature of WT and Tln13mut mice. To this end, platelets were fluorescently labeled, and the time at which thrombi started to form and finally occluded the vessels was recorded by intravital microscopy. We found that while Tln13mut platelets showed a tendency of delayed attachment to the injured vessel wall, Tln13mut mice showed a significantly prolonged vessel occlusion time in both venules (Figure 2B-C) and arterioles (Figure 2D-E) compared with control mice (Figure 2F). Collectively, our observations clearly indicate a critical role for direct Rap1/Talin1 interaction in regulating platelet function in vivo.
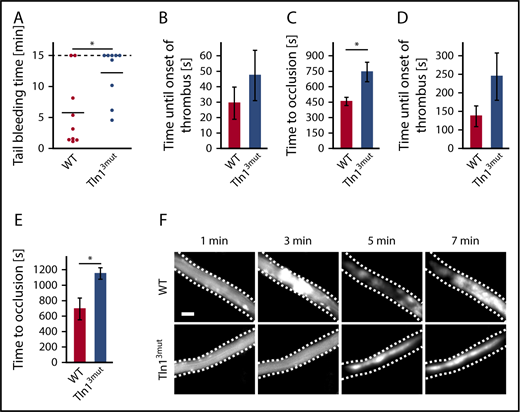
Hemostasis and thrombosis are reduced in Tln13mutmice. (A) Tail-bleeding time of WT and Tln13mut mice (cutoff at 15 minutes; n = 9). (B-C) Onset of thrombus formation (B) and vessel occlusion (C) measured in cremaster muscle venules of WT and Tln13mut mice after induction of phototoxic injury (n = 5). (D-E) Thrombus onset (D) and occlusion time (E) in injured cremaster muscle arterioles of WT and Tln13mut mice (n = 4). (F) Representative images of WT and Tln13mut cremaster muscle venules showing the progress of thrombus formation after 1, 3, 5, and 7 minutes. Scale bar, 25 µm. Values are given as means ± standard errors of the mean; n represents the number of mice. *P < .05.
Hemostasis and thrombosis are reduced in Tln13mutmice. (A) Tail-bleeding time of WT and Tln13mut mice (cutoff at 15 minutes; n = 9). (B-C) Onset of thrombus formation (B) and vessel occlusion (C) measured in cremaster muscle venules of WT and Tln13mut mice after induction of phototoxic injury (n = 5). (D-E) Thrombus onset (D) and occlusion time (E) in injured cremaster muscle arterioles of WT and Tln13mut mice (n = 4). (F) Representative images of WT and Tln13mut cremaster muscle venules showing the progress of thrombus formation after 1, 3, 5, and 7 minutes. Scale bar, 25 µm. Values are given as means ± standard errors of the mean; n represents the number of mice. *P < .05.
Rap1/Talin1 interaction controls integrin activity in neutrophils
Despite the important role of the Rap1/RIAM/Talin complex in leukocyte integrin activation, we still wondered whether the loss of Talin1/Rap1 interaction also impairs leukocyte integrin functions. Therefore, we isolated PMNs and performed adhesion assays in the presence and absence of TNF-α or phorbol-12-myristate-13-acetate (PMA). Unexpectedly, our assays revealed that adhesion of Tln13mut PMNs to the β2 integrin ligand ICAM-1, the α4β1 integrin ligand vascular cell adhesion molecule 1 (VCAM-1), and fibronectin, which is bound by several integrins, was significantly reduced compared with WT controls (Figure 3A). Importantly, intracellular expression of Talin1 and other integrin regulatory proteins, such as Rap1, Kindlin-3, and RIAM, and surface integrin levels were comparable between WT and mutant PMNs (Figure 3B,D). To test whether the reduced adhesion of Tln13mut PMNs was due to a defect in adhesion strengthening, we isolated PMNs from the bone marrow of control and Tln13mut mice, seeded them on ICAM-1–coated flow chambers, activated them with TNF-α, and applied a stepwise increase in shear stress within the chamber (detachment assay). In line with the results of the static adhesion assay, we observed an initial drop in the number of adherent Tln13mut cells when flow was started. However, increase in shear stress did not lead to further detachment of Tln13mut cells, indicating that direct Rap1 binding to Talin1 is not required for adhesion strengthening (Figure 3C). To investigate whether the loss of Rap1/Talin1 interaction also impairs other β2 integrin–mediated functions besides adhesion, we measured phagocytosis of serum-opsonized bacteria and found a significantly reduced bacterial uptake by Tln13mut PMNs compared with control PMNs (Figure 3E). Taken together, these data indicate that direct Rap1/Talin1 interaction is also critically involved in regulating neutrophil integrin functions.
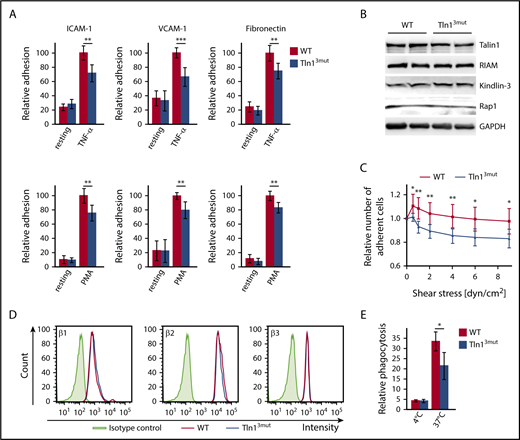
Integrin-mediated functions are impaired in Tln13mutPMNs. (A) Static adhesion of WT and Tln13mut PMNs on fibronectin, ICAM-1, and VCAM-1 upon stimulation with TNF-α or PMA compared with resting PMNs (n = 12 mice). (B) Analysis of Talin1, RIAM, Kindlin-3, and Rap1 expression in control and Tln13mut PMNs by western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading control. (C) Adhesion strengthening of TNF-α–stimulated WT and Tln13mut PMNs on ICAM-1 under increasing shear stress (0.5-9.0 dyn/cm2) measured as the number of adherent cells relative to initially adherent cells (n = 16 channels analyzed in 4 independent experiments). (D) Representative fluorescence-activated cell sorter blots illustrating surface levels of integrin β1, β2, and β3 on Gr-1+ neutrophils isolated from bone marrow. (E) Relative amount of fluorescently labeled Escherichia coli particles phagocytosed by WT and Tln13mut neutrophils (n = 3 mice). All values are given as means ± 95% confidence intervals. *P < .05, **P < .01, ***P < .001.
Integrin-mediated functions are impaired in Tln13mutPMNs. (A) Static adhesion of WT and Tln13mut PMNs on fibronectin, ICAM-1, and VCAM-1 upon stimulation with TNF-α or PMA compared with resting PMNs (n = 12 mice). (B) Analysis of Talin1, RIAM, Kindlin-3, and Rap1 expression in control and Tln13mut PMNs by western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading control. (C) Adhesion strengthening of TNF-α–stimulated WT and Tln13mut PMNs on ICAM-1 under increasing shear stress (0.5-9.0 dyn/cm2) measured as the number of adherent cells relative to initially adherent cells (n = 16 channels analyzed in 4 independent experiments). (D) Representative fluorescence-activated cell sorter blots illustrating surface levels of integrin β1, β2, and β3 on Gr-1+ neutrophils isolated from bone marrow. (E) Relative amount of fluorescently labeled Escherichia coli particles phagocytosed by WT and Tln13mut neutrophils (n = 3 mice). All values are given as means ± 95% confidence intervals. *P < .05, **P < .01, ***P < .001.
Direct Rap1/Talin1 interaction is required for neutrophil adhesion and extravasation in vivo
Next, we tested whether Tln13mut PMNs displayed a defect in response to inflammation in vivo. To do so, we investigated the adhesion and extravasation of PMNs on TNF-α–stimulated cremaster muscle venules using intravital microscopy. Although mean leukocyte rolling velocity was similar between control and Tln13mut neutrophils (Figure 4A), the adhesion efficiency (calculated as number of adherent PMNs per mm2 of vascular surface area divided by the systemic PMN count) of Tln13mut PMNs was significantly reduced compared with controls (Figure 4B), which is consistent with the reduced adhesion observed in isolated Tln13mut PMNs (Figure 3A). Importantly, we observed a strongly reduced number of extravasated PMNs in the perivascular cremaster muscle tissue of Tln13mut mice, suggesting that neutrophil recruitment into inflamed tissue is significantly impaired in the absence of direct Rap1 binding to Talin1 (Figure 4C-D). Our data therefore reveal a previously unappreciated role of the Rap1/Talin1 pathway in regulating leukocyte function in vivo.
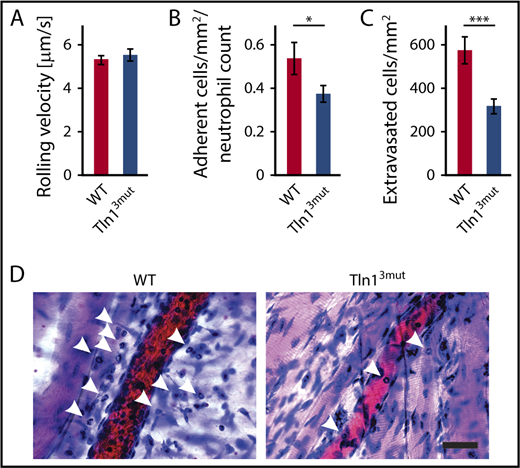
Neutrophil recruitment is reduced in Tln13mutmice. (A-D) In vivo leukocyte rolling velocity, adhesion efficiency, and extravasation were analyzed in mouse cremaster muscle venules of WT and Tln13mut mice 2 hours after intrascrotal injection of TNF-α. Rolling velocity (A) and adhesion efficiency (B) (adherent cells per mm2 normalized to the total neutrophil count) were analyzed using intravital microscopy. (C) Leukocyte extravasation was assessed in the perivascular region upon Giemsa staining of cremaster muscle whole mounts (n = 17 vessels in 4 WT and 4 Tln3mut mice). Values are given as means ± standard errors of the mean. (D) Representative images of Giemsa-stained whole mounts of WT and Tln13mut mice. Arrows point to extravasated neutrophils. Scale bar, 30 µm. *P < .05, ***P < .001.
Neutrophil recruitment is reduced in Tln13mutmice. (A-D) In vivo leukocyte rolling velocity, adhesion efficiency, and extravasation were analyzed in mouse cremaster muscle venules of WT and Tln13mut mice 2 hours after intrascrotal injection of TNF-α. Rolling velocity (A) and adhesion efficiency (B) (adherent cells per mm2 normalized to the total neutrophil count) were analyzed using intravital microscopy. (C) Leukocyte extravasation was assessed in the perivascular region upon Giemsa staining of cremaster muscle whole mounts (n = 17 vessels in 4 WT and 4 Tln3mut mice). Values are given as means ± standard errors of the mean. (D) Representative images of Giemsa-stained whole mounts of WT and Tln13mut mice. Arrows point to extravasated neutrophils. Scale bar, 30 µm. *P < .05, ***P < .001.
Direct Rap1/Talin1 interaction is not required for macrophage adhesion or spreading despite reduced integrin activation
The functional defects observed in platelets and neutrophils of Tln13mut mice suggest that Rap1/Talin1 interaction is crucial for circulating blood cells, which require rapid and dynamic integrin-mediated responses. We then asked whether the adhesion and spreading of other blood cells such as macrophages are affected in Tln13mut mice. In contrast to platelets and neutrophils, which respond immediately to vascular damage and infections, respectively, most macrophages differentiate within the tissue from extravasated monocytes and act as professional phagocytes and regulators of immune responses. We hypothesized that their functions are less dependent on fast integrin activation regulated by Rap1/Talin1 interaction. Therefore, we generated bone marrow–derived macrophages from WT and Tln13mut mice, which revealed normal expression of integrin regulatory proteins and similar levels of β1, β2, and β3 integrins on their cell surfaces (Figure 5A-B). Interestingly, adhesion and spreading of Tln13mut macrophages on β1, β2, and β3 integrin ligands were not altered compared with WT macrophages (Figure 5C-D), even though 9EG7 staining on Tln13mut macrophages revealed reduced levels of active β1 integrins (Figure 5E-F). These data suggest that in macrophages, unlike platelets and neutrophils, the activation of a reduced number of integrins is still sufficient to promote efficient cell adhesion and spreading.
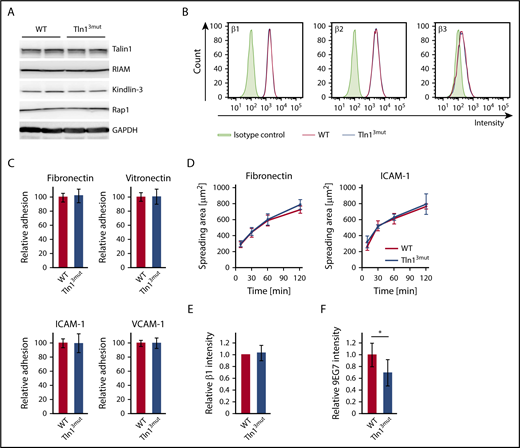
Adhesion and spreading of Tln13mutmacrophages are not impaired despite reduced levels of integrin activation. (A) Expression levels of Talin1, RIAM, Kindlin-3, and Rap1 in WT and Tln13mut macrophages assessed by western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading control. (B) Surface expression of integrin β1, β2, and β3 of WT (red) and Tln13mut (blue) macrophages shown in representative FACS blots. (C) Static adhesion of Tln13mut compared with WT macrophages on fibronectin, vitronectin, ICAM-1, and VCAM-1 (n = 6 experiments; cells isolated from 3 mice per genotype). (D) Spreading of WT and Tln13mut macrophages on fibronectin and ICAM-1 (n = 6 experiments; cells isolated from 3 mice per genotype). (E-F) Quantification of total (E) and 9EG7+ (F) β1 integrin staining intensity in regions of clustered integrins in WT and Tln13mut macrophages 2 hours after plating on a fibronectin-coated surface (WT values for each experiment set to 1; n = 6 experiments; cells from 2 mice of each genotype). Values are given as means ± 95% confidence intervals. *P < .05.
Adhesion and spreading of Tln13mutmacrophages are not impaired despite reduced levels of integrin activation. (A) Expression levels of Talin1, RIAM, Kindlin-3, and Rap1 in WT and Tln13mut macrophages assessed by western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading control. (B) Surface expression of integrin β1, β2, and β3 of WT (red) and Tln13mut (blue) macrophages shown in representative FACS blots. (C) Static adhesion of Tln13mut compared with WT macrophages on fibronectin, vitronectin, ICAM-1, and VCAM-1 (n = 6 experiments; cells isolated from 3 mice per genotype). (D) Spreading of WT and Tln13mut macrophages on fibronectin and ICAM-1 (n = 6 experiments; cells isolated from 3 mice per genotype). (E-F) Quantification of total (E) and 9EG7+ (F) β1 integrin staining intensity in regions of clustered integrins in WT and Tln13mut macrophages 2 hours after plating on a fibronectin-coated surface (WT values for each experiment set to 1; n = 6 experiments; cells from 2 mice of each genotype). Values are given as means ± 95% confidence intervals. *P < .05.
Discussion
In this study, we generated Rap1 binding–deficient Talin1 knockin mice to investigate the physiological importance of direct Rap1/Talin1 interaction in integrin signaling. Tln13mut mice develop normally and are viable and apparently healthy. Although MEFs isolated from Tln13mut mice show normal integrin function, likely because of functional compensation by Talin2, hematopoietic cells such as platelets and neutrophils, which express no or low levels of Talin2, showed significant defects in integrin activation and integrin-dependent cell functions. Our study thus for the first time demonstrates the important role of Rap1/Talin1 interaction in vivo.
One important finding of our study is that platelet integrin activity, aggregation, and spreading as well as hemostasis and thrombosis are impaired in Rap1 binding–deficient Tln13mut mice, demonstrating the critical physiological relevance of this interaction in vivo. The platelet defects and the resulting phenotype in Tln13mut mice are similar to mice lacking the most prominent Rap1 isoform (Rap1b) in platelets, which exhibit moderate integrin activation and signaling defects.7 A recent elegant study showed that the deletion of both Rap1 genes (Rap1a and Rap1b) in the megakaryocyte lineage26 led to a more severe defect in integrin activation, where ∼80% of the platelet integrins remained inactive upon activation via different agonists. Although Talin1_3mut protein likely fails to interact with both Rap1 proteins due to their high similarity,14 Tln13mut mice indeed showed a milder phenotype. There are several factors that may contribute to this difference. First, the interaction between Rap1 and mutant Talin1 is impaired but may not be completely abolished in cells. This possibility is supported by the rather moderate cell adhesion, spreading, and Talin1 recruitment defects in cells expressing double- or triple-mutant Talin1 proteins compared with Talin1 mutant lacking the complete F0 domain (supplemental Figure 2D-F). That is, residual interaction between Rap1 and Talin1 mutant may still support some Talin1 recruitment to regulate integrin activation and signaling, especially in platelets, which express high protein levels of Rap1b and Talin1.24 Such a possibility is also consistent with the more severe phenotypes observed in our Tln13mut mice compared with the recently reported Talin1 (R35E) knockin mice23 because of the less disrupted Rap1/Talin1 interaction of the latter (supplemental Figures 3A-D and 4A-F). Second, abolishing Rap1/Talin1 interaction is fundamentally different from complete Rap1a/Rap1b double knockout. The former blocks only 1 of many Rap1 functions, in contrast to the latter. It is well known that the small GTPase Rap1 serves as a central signaling unit and regulates many integrin-dependent (Rap1 is involved in both integrin inside-out and outside-in signaling27 ) and -independent signaling pathways by interacting with a variety of effector proteins; complete loss of Rap1a/Rap1b expression has therefore much more dramatic functional consequences. Third, despite the low RIAM expression in platelets, the formation of some Rap1/RIAM/Talin1 complexes may partially compensate for deficient Rap1/Talin1 interaction to regulate Talin recruitment. Additional depletion of RIAM in Talin1 (R35E) knockin mice did not lead to a more obvious platelet phenotype.23 However, functional compensation by RIAM might be dispensable, given that Talin1 (R35E) platelets alone have very mild defects. Therefore, generating Tln13mut mice with further depletion of RIAM instead will clarify this.
We would also like to mention that residual (20%) platelet integrin activity was observed even in Rap1a/b double-mutant platelets,26 suggesting the involvement of Rap1-independent pathways (eg, PIP2-mediated Talin recruitment)28,29 in regulating basal platelet function. Nevertheless, our study clearly showed significant platelet defects when the direct Rap1/Talin1 pathway was impaired and demonstrated its important role in regulating platelet integrin activity and function in vivo.
A second important finding of our study is that the direct Rap1/Talin1 pathway is also used in neutrophils to regulate integrin activity. Tln13mut neutrophils revealed impaired β2 integrin–mediated adhesion and phagocytosis, indicating that direct Rap1/Talin interaction is indispensable for full β2 integrin function. This observation is surprising, because we and others have shown that leukocyte β2 integrin functions are critically dependent on the Rap1/RIAM/Talin1 complex.6,10 Moreover, Tln13mut neutrophils also exhibited defective adhesion to β1 and β3 integrin ligands, suggesting that direct Rap1/Talin1 interaction is of general importance to regulate the activity of all major integrin classes, whereas Rap1/RIAM/Talin1 is mainly involved in β2 integrin activation. Notably, both Tln13mut and RIAM knockout mice show defective β2 integrin–mediated neutrophil adhesion and extravasation into inflamed tissue, indicating the synergistic action of both Rap1/Talin1 and Rap1/RIAM/Talin1 pathways in regulating neutrophil integrin signaling. However, we did not observe clear leukocytosis in Tln3mut mice, which is present in RIAM knockout mice, indicating that the RIAM-dependent pathway plays a major role in β2 integrin–mediated leukocyte extravasation. Moreover, Tln13mut macrophages exhibited no adhesion or spreading defect despite reduced β1 integrin activity, suggesting that direct Rap1/Talin1 interaction is particularly important in circulating cells, like platelets and neutrophils, which require fast and dynamic integrin-mediated responses. Additional studies are required to address this point. Nevertheless, our data provide strong evidence that direct Rap1/Talin1 interaction is important for regulating integrin signaling in the physiological context.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Soo Jin Min-Weißenhorn and members of the transgenic facility team of the Max Planck Institute of Biochemistry for help in generating the Tln13mut mice. The authors also thank Reinhard Fässler for his support and Arnoud Sonnenberg for critically reading the manuscript.
The work was supported by the Deutsche Forschungsgemeinschaft (SFB914 TP A01, B01, B03), the Max Planck Society, and grant R01GM62823 from National Institute of General Medical Sciences, National Institutes of Health.
Authorship
Contribution: T.B., S.K., I.R., L.Z., L.M., and M.M. designed and performed the experiments and analyzed data; C.A.R., M.S., and J.Q. designed experiments and analyzed data; M.M. supervised the work; and J.Q. and M.M. wrote the paper with input from all other authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Markus Moser, Max Planck Institute of Biochemistry, Department of Molecular Medicine, Am Klopferspitz 18, D-82152 Martinsried, Germany; e-mail: moser@biochem.mpg.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal