

The classic Philadelphia chromosome (Ph)–negative myeloproliferative neoplasms (MPNs) are a heterogeneous group of hematopoietic stem-cell diseases, characterized by activated JAK/STAT signaling and significant phenotypic mimicry, including a propensity for evolution to myeloid blast phase disease. Effective therapeutic options are limited for patients with Ph− MPNs in the blast phase (MPN-BP), and allogeneic stem-cell transplantation is the only known cure. Our increasing understanding of the molecular pathogenesis of this group of diseases, coupled with the increasing availability of targeted agents, has the potential to inform new subset-specific therapeutic approaches. Ultimately, progress in MPN-BP will hinge on prospective clinical and translational investigations with the goal of generating more effective treatment interventions. This case-based review highlights the molecular and clinical heterogeneities of MPN-BP and incorporates a treatment algorithm that underscores the importance of a personalized approach to this challenging group of diseases.
Introduction
The classic Philadelphia chromosome (Ph)–negative myeloproliferative neoplasms (MPNs), including essential thrombocythemia (ET), polycythemia vera (PV), and primary myelofibrosis (PMF), are a heterogeneous group of hematopoietic stem-cell diseases characterized by driver mutations in JAK2, CALR, and MPL genes that result in activation of the JAK/STAT signaling pathway.1,-3 Phenotypically, these diseases share a variable propensity to thrombovascular complications, constitutional symptoms, overt myelofibrotic transformation, and evolution to a myeloid blast phase.4 The terms PMF in blast phase and post-PV/ET myelofibrosis (MF) in blast phase were coined by the International Working Group for Myelofibrosis Research and Treatment in recognition of the significant clinical and biologic differences between acute leukemia evolving from the Ph− MPNs and acute myeloid leukemia (AML) arising de novo.5 Collectively, leukemic transformation from the Ph− MPNs, defined as ≥20% myeloblasts in the peripheral blood and/or bone marrow in an individual with a preexisting MPN, is often referred to as MPN blast phase (MPN-BP).6
There is no standard approach to the management of MPN-BP, and outcomes are generally poor. The median survival is in the 3- to 5-month range in published retrospective series, even in the context of intensive induction chemotherapy.7,,,-11 Allogeneic stem-cell transplantation (SCT) is the only approach reported to have the potential to significantly change the natural history of MPN-BP, but applicability has historically been limited by the refractory nature of these diseases and the older age (median age is in the mid 60s to early 70s) of patients at the time of presentation.
Despite these disheartening outcomes, there is reason for a renewed sense of optimism based on our increasing understanding of the biologic mechanisms that drive these disparate group of disorders, as well as the increasing availability of new agents and approaches. In this case-based review, I will highlight a few cases that illustrate contemporary therapeutic strategies and challenges in this otherwise poor-prognosis group of diseases.
Who is at the highest risk of transformation to MPN-BP?
Given the historically poor outcomes associated with MPN-BP, identification of patients with Ph− MPNs at high risk of myeloid blast phase transformation could facilitate early intervention, such as allogeneic SCT. Risk factors associated with leukemic transformation (Table 1), include clinicopathologic12,,,,,,,,,,,,-25 and cytogenetic or molecular/genetic risk factors.18,26,,,,,,-33 The predictive ability of these factors for evolution to MPN-BP is variable and limited by the heterogeneity of disease features, coupled with the retrospective nature of several published series.34
Risk factors for evolution to MPN-BP
| Risk factor | Comments | Reference |
|---|---|---|
| Clinicopathologic | Cumulative incidence >20 y for MPN-BP: 3.8%, 6.8%, and 14.2% in ET, PV, and PMF, respectively, in contemporary series from the Mayo clinic; evolution to overt MF phenotype often precedes MPN-BF in PV; in post-PV MF, high circulating CD34 counts; platelet count <100 × 109/L implicated in MPN-BP; median age at diagnosis: 61, 60, and 65 y for PV, ET, and MF, respectively | 12,15,16,19,33,95,96 |
| MPN subtype | ||
| PMF | ||
| Post-ET MF | ||
| Post-PV MF | ||
| Cytoreductive agents | Controversy regarding leukemogenic potential of hydroxyurea in the MPNs, which is not supported by evidence from large retrospective series | 15,19,,,,-24 |
| Phosphorus-32 | ||
| Chlorambucil | ||
| Piprobroman | ||
| Busulphan | ||
| Laboratory parameters | Blasts ≥3% + platelets <100 × 109/L indicates high risk for MPN-BP; blasts ≥10% + platelets <50 × 109/L indicates accelerated disease/very high risk for MPN-BP; transfusion dependency; WBC >30 × 109/L; WBC >15 × 109/L + age >61 y + abnormal karyotype (in PV) | 13,14,17,-19 |
| Circulating blasts | ||
| Anemia | ||
| Leukocytosis | ||
| Thrombocytopenia | ||
| Molecular/genetic | Unfavorable karyotype in DIPSS-plus: +8, −7/7q, i(17q), −5/5q−, 12p−, inv(3), and 11q23 or chromosome 5, 7, or 17p abnormalities indicates >6× risk of MPN-BP | 18,26,27,29,,-32 |
| Unfavorable karyotype (PMF) | ||
| 17p deletion | ||
| Mutations | ||
| ASXL1, IDH1/2, EZH2, SRSF2 | ||
| TET2, TP53 | ||
| Absence of CALR (in PMF) | ||
| ≥2 mutations | ||
| Germ line duplication of ATG2B/GSKIP* | ||
| Prognostic scoring systems (PMF) | In DIPSS, high risk for evolution to MPN-BP indicated by 7.8 for intermediate-2–risk disease and 24.9 for high-risk disease when compared with low risk; in DIPSS-plus, unfavorable karyotype + platelet count <100 000/uK indicates high risk for MPN-BP; MIPSS70 very high-risk category: 23% developed MPN-BP (HR, 13.3) when compared with low risk | 25,28 |
| Higher-risk DIPSS score | ||
| DIPSS-plus score | ||
| High-risk MIPSS70 score | ||
| Very high-risk MIPSS70-plus score† |
| Risk factor | Comments | Reference |
|---|---|---|
| Clinicopathologic | Cumulative incidence >20 y for MPN-BP: 3.8%, 6.8%, and 14.2% in ET, PV, and PMF, respectively, in contemporary series from the Mayo clinic; evolution to overt MF phenotype often precedes MPN-BF in PV; in post-PV MF, high circulating CD34 counts; platelet count <100 × 109/L implicated in MPN-BP; median age at diagnosis: 61, 60, and 65 y for PV, ET, and MF, respectively | 12,15,16,19,33,95,96 |
| MPN subtype | ||
| PMF | ||
| Post-ET MF | ||
| Post-PV MF | ||
| Cytoreductive agents | Controversy regarding leukemogenic potential of hydroxyurea in the MPNs, which is not supported by evidence from large retrospective series | 15,19,,,,-24 |
| Phosphorus-32 | ||
| Chlorambucil | ||
| Piprobroman | ||
| Busulphan | ||
| Laboratory parameters | Blasts ≥3% + platelets <100 × 109/L indicates high risk for MPN-BP; blasts ≥10% + platelets <50 × 109/L indicates accelerated disease/very high risk for MPN-BP; transfusion dependency; WBC >30 × 109/L; WBC >15 × 109/L + age >61 y + abnormal karyotype (in PV) | 13,14,17,-19 |
| Circulating blasts | ||
| Anemia | ||
| Leukocytosis | ||
| Thrombocytopenia | ||
| Molecular/genetic | Unfavorable karyotype in DIPSS-plus: +8, −7/7q, i(17q), −5/5q−, 12p−, inv(3), and 11q23 or chromosome 5, 7, or 17p abnormalities indicates >6× risk of MPN-BP | 18,26,27,29,,-32 |
| Unfavorable karyotype (PMF) | ||
| 17p deletion | ||
| Mutations | ||
| ASXL1, IDH1/2, EZH2, SRSF2 | ||
| TET2, TP53 | ||
| Absence of CALR (in PMF) | ||
| ≥2 mutations | ||
| Germ line duplication of ATG2B/GSKIP* | ||
| Prognostic scoring systems (PMF) | In DIPSS, high risk for evolution to MPN-BP indicated by 7.8 for intermediate-2–risk disease and 24.9 for high-risk disease when compared with low risk; in DIPSS-plus, unfavorable karyotype + platelet count <100 000/uK indicates high risk for MPN-BP; MIPSS70 very high-risk category: 23% developed MPN-BP (HR, 13.3) when compared with low risk | 25,28 |
| Higher-risk DIPSS score | ||
| DIPSS-plus score | ||
| High-risk MIPSS70 score | ||
| Very high-risk MIPSS70-plus score† |
DIPSS, Dynamic International Prognostic Scoring System; HR, hazard ratio; MIPSS, Mutation-Enhanced International Prognostic Score System; WBC, white blood cell.
Associated with a familial MPN phenotype with high risk of MF and MPN-BP evolution.
Unfavorable karyotype in MIPSS70-plus: any abnormal karyotype other than normal karyotype or sole abnormalities of 20q−, 13q−, +9, chromosome 1 translocation/duplication, or sex chromosome abnormality other than −Y.
In general, patients at high risk of MPN-BP (Table 1) in whom allogeneic transplantation should be considered include those with MF who have Dynamic International Prognostic Scoring System intermediate 2 or high-risk disease (there is an established consensus in this regard), unfavorable karyotype, increasing circulating blasts, transfusion dependency, or thrombocytopenia.35 Patients with accelerated-phase disease (MPN-AP), often simply defined as 10% to 19% blasts, have a median survival of <2 years18,36 and merit consideration for treatment strategies similar to those employed for MPN-BP, including allogeneic SCT.
Recent studies have highlighted the importance of host genetic and environmental factors, hematopoietic stress, and aging in predisposition to clonal hematopoiesis and in facilitating clonal dominance and emergence of the MPN phenotype.31,37,-39 The evolutionary pathways leading to clonal expansion, including acquisition of additional deleterious mutations (Figure 1) that promote clonal evolution and progression toward MPN-BP, have recently been reviewed.1 The mutational profile of MPN-BP is distinct from that of AML arising de novo and is characterized by mutations in IDH1/2, TET2, SRSF2, ASXL1, and TP53.11,26,29,30,40,-42 By contrast, mutations in NPM1-FLT3, which are typical of AML de novo, are uncommon in MPN-BP. Retrospective analysis of paired samples obtained from patients with JAK2 V617F–mutant MPNs during the chronic phase and at the time of transformation to MPN-BP has demonstrated the likelihood of >1 evolutionary pathway to leukemogenesis. In some cases, as expected, the leukemia blasts demonstrated persistence of the JAK2 V617F clone. In other cases, however, JAK2-mutant MPNs evolved to a JAK2 wild-type clone in MPN-BP, suggesting the presence of a common JAK2 V617F− ancestral clone or the possibility that the MPN was biclonal from the outset.43,44
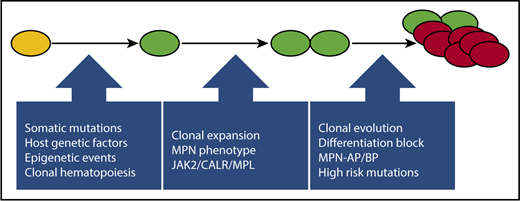
An evolutionary pathway to MPN-BP. After the acquisition of a JAK2 V617F or similar driver mutation by a hematopoietic stem cell (depicted in yellow), host genetic, epigenetic, and microenvironmental factors can facilitate the emergence of asymptomatic clonal hematopoiesis (depicted in green), subsequent clonal expansion, and emergence of an MPN phenotype (depicted as doublet of green cells). The acquisition of additional mutations, including high-risk mutations (ASXL1, IDH1/2, SRSF2, EZH2, TP53), may lead to clonal evolution and disease progression and may ultimately culminate in MPN-BP (depicted as an outgrowth/cluster of red cells).
An evolutionary pathway to MPN-BP. After the acquisition of a JAK2 V617F or similar driver mutation by a hematopoietic stem cell (depicted in yellow), host genetic, epigenetic, and microenvironmental factors can facilitate the emergence of asymptomatic clonal hematopoiesis (depicted in green), subsequent clonal expansion, and emergence of an MPN phenotype (depicted as doublet of green cells). The acquisition of additional mutations, including high-risk mutations (ASXL1, IDH1/2, SRSF2, EZH2, TP53), may lead to clonal evolution and disease progression and may ultimately culminate in MPN-BP (depicted as an outgrowth/cluster of red cells).
Treatment of MPN-BP: intensive chemotherapy
The therapeutic approaches in MPN-BP have ranged from supportive care to intensive chemotherapy and allogeneic SCT (Table 2). The published series are retrospective in nature, and there is significant heterogeneity with regard to the patient population involved, treatment strategies employed, and sample size. There is also a lack of standardization with regard to the criteria employed to evaluate response to therapy.6 These issues pose a significant challenge when trying to evaluate the relative contribution of specific therapeutic approaches. Patients treated with a supportive care–only approach had short median survival, in the 6-week to 2-month range.8,9,11,45
Treatment series focused on MPN-AP or MPN-BP
| Treatment approach | Patient population | Comments | Survival | Reference |
|---|---|---|---|---|
| IC ± allogeneic HCT | MPN-BP (n = 91) | 24 (26%) received IC; 41% of IC group had return to cMPN | OS, 2.6 mo; 3.9 mo in IC group; 1 patient underwent allogeneic HCT | 8 |
| MPN-BP (n = 74) | 41 (55%) received IC; CR/CRi, 46% | OS, 5 mo; OS, 6 mo and PFS, 5 mo with IC; 8 patients underwent early SCT; 73% of these were alive at median of 31 mo | 9 | |
| MPN-BP (n = 75) | 39 (51%) treated with curative intent and received IC and/or underwent allogeneic HCT | OS, 6.6 mo; median OS of curative intent group, 9.4 mo; 17 (23%) underwent allogeneic HCT; median OS, 47 mo for allogeneic HCT | 50 | |
| MPN-BP (n = 248) | 66 of 122 patients assessable for response received IC; 26 received HMA | OS, 3 mo; 1-y survival before y 2000, 5%; it was 20% in ≥2010; CR, 35% with IC; CR <5% with HMA | 10 | |
| HMAs | MPN-BP (n = 26); MPN-AP (n = 28) | ORR, 52% (CR, 24%) with azacitidine therapy; median response duration, 9 mo; recurrence of cMPN in 39% responders | OS, 11 mo | 67 |
| MPN-BP (n = 19) | ORR, 47% (CR, 26%) with azacitidine therapy | OS, 9.9 mo | 64 | |
| MPN-BP (n = 21); MPN-AP (n = 13); PMF chronic phase (n = 11) | ORR in MPN-BP, 29% with decitabine therapy; median response duration, 7 mo | OS, 6.9 mo in MPN-BP; OS, 9.7 mo in MPN-AP | 65 | |
| MPN-BP (n = 11) | 6 patients received decitabine; 5 patients underwent allogeneic HCT | 67% in decitabine group alive at 9 mo; 53% in transplantation group alive at 20 mo | 66 | |
| JAK inhibition | R/R AML (n = 38), including MPN-BP (n = 18) | 3 of 18 patients with MPN-BP treated with ruxolitinib at dose of 25 mg twice daily achieved CR/CRi | NR | 77 |
| R/R acute leukemia (n = 28), including 7 with antecedent Ph− MPN | 1 patient with AML (prior MDS) achieved CRp; no objective response in MPN-BP cohort; ruxolitinib dosed between 50 and 200 mg twice daily | NR | 78 | |
| JAK inhibition + HMA | MPN-AP/MPN-BP (n = 21) | Phase 1 study of ruxolitinib + decitabine; 7 of 21 responded | OS, 10.4 mo | 80 |
| MPN-BP (n = 10) | Phase 1/2 study of ruxolitinib + decitabine | NR | 79 | |
| Allogeneic HCT | MPN-BP (n = 43); MDS/MPN-BP (n = 17) | 60 underwent allogeneic HCT; 3-y TRM, 22%; 3-y CIR, 68% | OS, 18% at 3 y; 3-y LFS, 18% for allogeneic HCT in CR; 3-y LFS, 3% for allogeneic HCT in advanced disease | 47 |
| MPN-BP (n = 13) | 8 underwent allogeneic HCT; 5 of 8 were in CR or cMPN at time of allogeneic HCT | At median follow-up of 20 mo, 6 patients were alive in CR post–allogeneic HCT | 48 | |
| MF chronic phase (n = 41); MF-BP (n = 14) | Of 14 MF-BP patients who underwent allogeneic HCT, 7 survived; median follow-up, 31 mo | OS, 49% at 2 y in MF-BP cohort; 3 of 6 patients in remission and 4 of 8 patients with relapsed disease at allogeneic HCT were long-term survivors | 49 | |
| MF-BP (n = 46) | 1-y TRM, 28%, CIR at 3 y, 47%; only 8 of 46 were in CR pre–allogeneic HCT | 3-y PFS, 26%; OS, 33%; CR pre–allogeneic HCT was significantly predictive of OS and PFS | 46 |
| Treatment approach | Patient population | Comments | Survival | Reference |
|---|---|---|---|---|
| IC ± allogeneic HCT | MPN-BP (n = 91) | 24 (26%) received IC; 41% of IC group had return to cMPN | OS, 2.6 mo; 3.9 mo in IC group; 1 patient underwent allogeneic HCT | 8 |
| MPN-BP (n = 74) | 41 (55%) received IC; CR/CRi, 46% | OS, 5 mo; OS, 6 mo and PFS, 5 mo with IC; 8 patients underwent early SCT; 73% of these were alive at median of 31 mo | 9 | |
| MPN-BP (n = 75) | 39 (51%) treated with curative intent and received IC and/or underwent allogeneic HCT | OS, 6.6 mo; median OS of curative intent group, 9.4 mo; 17 (23%) underwent allogeneic HCT; median OS, 47 mo for allogeneic HCT | 50 | |
| MPN-BP (n = 248) | 66 of 122 patients assessable for response received IC; 26 received HMA | OS, 3 mo; 1-y survival before y 2000, 5%; it was 20% in ≥2010; CR, 35% with IC; CR <5% with HMA | 10 | |
| HMAs | MPN-BP (n = 26); MPN-AP (n = 28) | ORR, 52% (CR, 24%) with azacitidine therapy; median response duration, 9 mo; recurrence of cMPN in 39% responders | OS, 11 mo | 67 |
| MPN-BP (n = 19) | ORR, 47% (CR, 26%) with azacitidine therapy | OS, 9.9 mo | 64 | |
| MPN-BP (n = 21); MPN-AP (n = 13); PMF chronic phase (n = 11) | ORR in MPN-BP, 29% with decitabine therapy; median response duration, 7 mo | OS, 6.9 mo in MPN-BP; OS, 9.7 mo in MPN-AP | 65 | |
| MPN-BP (n = 11) | 6 patients received decitabine; 5 patients underwent allogeneic HCT | 67% in decitabine group alive at 9 mo; 53% in transplantation group alive at 20 mo | 66 | |
| JAK inhibition | R/R AML (n = 38), including MPN-BP (n = 18) | 3 of 18 patients with MPN-BP treated with ruxolitinib at dose of 25 mg twice daily achieved CR/CRi | NR | 77 |
| R/R acute leukemia (n = 28), including 7 with antecedent Ph− MPN | 1 patient with AML (prior MDS) achieved CRp; no objective response in MPN-BP cohort; ruxolitinib dosed between 50 and 200 mg twice daily | NR | 78 | |
| JAK inhibition + HMA | MPN-AP/MPN-BP (n = 21) | Phase 1 study of ruxolitinib + decitabine; 7 of 21 responded | OS, 10.4 mo | 80 |
| MPN-BP (n = 10) | Phase 1/2 study of ruxolitinib + decitabine | NR | 79 | |
| Allogeneic HCT | MPN-BP (n = 43); MDS/MPN-BP (n = 17) | 60 underwent allogeneic HCT; 3-y TRM, 22%; 3-y CIR, 68% | OS, 18% at 3 y; 3-y LFS, 18% for allogeneic HCT in CR; 3-y LFS, 3% for allogeneic HCT in advanced disease | 47 |
| MPN-BP (n = 13) | 8 underwent allogeneic HCT; 5 of 8 were in CR or cMPN at time of allogeneic HCT | At median follow-up of 20 mo, 6 patients were alive in CR post–allogeneic HCT | 48 | |
| MF chronic phase (n = 41); MF-BP (n = 14) | Of 14 MF-BP patients who underwent allogeneic HCT, 7 survived; median follow-up, 31 mo | OS, 49% at 2 y in MF-BP cohort; 3 of 6 patients in remission and 4 of 8 patients with relapsed disease at allogeneic HCT were long-term survivors | 49 | |
| MF-BP (n = 46) | 1-y TRM, 28%, CIR at 3 y, 47%; only 8 of 46 were in CR pre–allogeneic HCT | 3-y PFS, 26%; OS, 33%; CR pre–allogeneic HCT was significantly predictive of OS and PFS | 46 |
CIR, cumulative incidence of relapse; cMPN, reversion to chronic phase MPN; CR, complete response; CRi, CR with incomplete hematologic recovery; HCT, hematopoietic cell transplantation; HMA, hypomethylating agent; IC, intensive chemotherapy; NR, not reported; ORR, overall response rate; OS, overall survival; PFS, progression-free survival; R/R, relapsed/refractory; TRM, transplantation-related mortality.
Patient 1
A 64-year-old man was diagnosed with ET 21 years before, in the context of an evaluation of signs and symptoms consistent with a transient ischemic attack. He was treated with hydroxyurea in conjunction with low-dose aspirin. Nineteen years later, he presented with complaints of fatigue and exertional dyspnea. The complete blood count (CBC) revealed a WBC count of 6.5 × 109/L, hemoglobin of 85 g/L, and platelet count of 613 × 109/L. The WBC differential revealed 30% circulating blasts. The bone marrow biopsy revealed a hypercellular marrow with 40% blasts, abundant megakaryocytes, and focally increased fibrosis. Immunophenotyping revealed that the blasts were CD34+, CD33+, HLA-DR+, and CD117+. The bone marrow cytogenetic analysis revealed a complex karyotype: 46, XY[55%]; 42, XY, ins(2;5)(q31;p13p15), del(4)(q27q35), −5, −6, add(7)(p15), dic(8;19)(p11.1;p12), −22, +der(?)t(/;6)(?;q12)[5%]; clone 2: 41, idem, −16, add(21)(q22)[20%]; clone 3:42, idem, add(16)9q22)[20%]. Molecular analysis using an institutional next-generation sequencing (NGS) panel (limit of detection, 10% mutant alleles) revealed the following pathogenic variants: JAK2 V617F, ASXL1 Q910Tfs*14, and IDH2 R140Q. The patient had been working full time and exercising regularly, without any limitations, before this presentation.
On the basis of the patient’s overall fitness level and the intent to proceed to allogeneic SCT as soon as disease control was achieved, induction chemotherapy was initiated with high-dose cytarabine (3 g/m2; days 1 and 5) and mitoxantrone (30 mg/m2; days 1 and 5) according to a regimen commonly used at our institution for high-risk AML.51,52 The nadir bone marrow biopsy at day 14 showed persistent leukemia. His performance status remained excellent, and a second induction attempt with high-dose cytarabine and etoposide was administered. The subsequent bone marrow biopsy at day 14 of the second induction also showed persistent leukemia (40% cellular, 50% blasts).
At this point, given the presence of IDH2 R140Q and the availability of a clinical trial focused on patients with late-stage AML harboring an IDH2 mutation, participation in the clinical trial was recommended. The patient was subsequently enrolled in a clinical trial of enasidenib for late-stage AML and was randomized to the experimental arm of the study. Therapy with enasidenib was initiated at 100 mg per day administered orally in 28-day cycles. His bone marrow biopsy after 2 cycles showed a hypercellular marrow, with a decline in blasts to 9%. The bone marrow cytogenetic analysis revealed a reversion to a normal male karyotype. The most recent bone marrow biopsy at the 2-year time point was mildly hypercellular (50% to 60% cellular), with increased granulopoiesis and markedly atypical megakaryocytic proliferation, with a marginal increase in blasts at 3% to 4%. His recent blood counts revealed the following: WBC count of 15 × 109/L, 89% neutrophils, 2% blasts, hemoglobin of 139 g/L, and platelet count of 200 × 109/L. Interestingly, recent NGS analysis revealed the following pathogenic variants: JAK2 V617F and ASXL1 Q910Tfs*14. The IDH2 mutation was not detected (sensitivity of NGS assay, 10% mutant alleles).
StatusThe patient is 2.5 years out from the original diagnosis of MPN-BP and is working full time. His performance status is excellent, and he is exercising on a near-daily basis. He remains on 100 mg of enasidenib daily and 81 mg of low-dose aspirin daily. An allogeneic SCT evaluation has been recommended, but the patient has elected not to proceed with transplantation at this time.
Late-stage MPN-BP associated with IDH1/2 mutations
The advent of therapeutic agents targeted to mutant IDH1/2 enzymes has provided a new treatment approach for patients with high-risk myeloid neoplasms associated with IDH2 mutations. IDH2 mutations are present at a low frequency, in the 2% to 4% range, in the chronic phase of Ph− MPNs.32,41,53 The incidence increases with evolution to MPN-BP and is in the 13% to 31% range in published retrospective series.11,26,40,41 Wild-type IDH1/2 enzymes catalyze the conversion of isocitrate to α-ketoglutarate. Mutant IDH1/2 results in conversion of isocitrate to R-2-hydoxyglutarate (2-HG), an oncometabolite that competitively inhibits α-ketoglutarate–dependent enzymes, including TET2 and the Jumanji enzymes, resulting in DNA and histone hypermethylation, epigenetic dysregulation, and differentiation block.54
Enasidenib (AG-221) and ivosidenib (AG-120) are small-molecule inhibitors of mutant IDH2 and IDH1, respectively, which bind the catalytically active sites, preventing conversion of isocitrate to 2-HG, reversing the epigenetic dysregulation, and facilitating cellular differentiation. These agents were associated with significant clinical activity in early-phase trials of relapsed or refractory IDH1/2-mutant AML.55,-57 2-HG levels were uniformly downregulated and served as a useful pharmacodynamic marker in early-phase development of these agents, but decline in 2-HG levels did not correlate with response. The overall response rate with enasidenib in relapsed or refractory AML was 40% (CR/CRi rate of ∼26%), and more than half of patients enrolled had received ≥2 prior AML-directed regimens. The median overall survival was 9.3 months, but in those who had a CR or partial response, the median survival was 19.7 months. The agent was well tolerated, with the most frequent adverse events being indirect hyperbilirubinemia and IDH differentiation syndrome.58
On the basis of these encouraging results, enasidenib and ivosidenib were recently approved in the United States by the US Food and Drug Administration for relapsed or refractory AML associated with an IDH2 or IDH1 mutation, respectively. Enasidenib and ivosidenib are therefore potential options for patients with MPN-BP associated with IDH1/2 mutations who are refractory to or have relapsed after prior therapy. There is a paucity of information, however, with regard to both clinical trial55,-57 and real-world experiences with these agents in MPN-BP. Enrollment in studies that aim to validate the potential promise of this approach in MPN-BP remains the preferred option. Currently, there are ongoing trials in which enasidenib and ivosidenib are being combined with azacitidine or intensive induction chemotherapy in the frontline setting in AML, including AML arising from prior Ph− MPNs. Interestingly, in a murine model of combined JAK2/IDH2-mutant MPNs, the combinations of JAK inhibition with ruxolitinib and IDH2 inhibition with enasidenib reduced disease burden to a greater extent than was seen with JAK inhibition alone. Similar cooperativity with combined JAK/IDH2 inhibition was seen in primary cells from MPN patients with both mutations, suggesting the potential promise of this approach.59 Such combinations require validation in the context of prospective clinical trials.
Patient 2
MPN-BP associated with TP53 mutations
A 77-year-old man was diagnosed with PV 9 years ago. Phlebotomy and low-dose aspirin were initiated, followed by hydroxyurea, in line with treatment recommendations for upfront therapy for high-risk PV.60
Eight years after the original diagnosis of PV, he presented with significant fatigue. His performance status had declined significantly to an Eastern Cooperative Oncology Group performance status of 3. CBC revealed a platelet count of 110 × 109/L, WBC count of 6.7 × 109/L, and hemoglobin of 104 g/L. The peripheral blood smear was notable for leukoerythroblastosis, occasional hypogranular neutrophils, and 15% circulating blasts. The bone marrow biopsy revealed a hypercellular marrow with panmyelosis and marked reticulin (MF grade 3) and collagen fibrosis, with CD34+ blasts comprising 20% to 30% of the overall cellularity (Figure 2). The cytogenetic analysis performed on the peripheral blood revealed a complex karyotype, including abnormalities of chromosomes 5 and 7 and trisomy 8. Pathogenic variants detected on NGS analysis included JAK2 V617F, TP53 H168R, and TP53 S215G. Serum lactate dehydrogenase (LDH) was significantly elevated at 2930 IU/L.
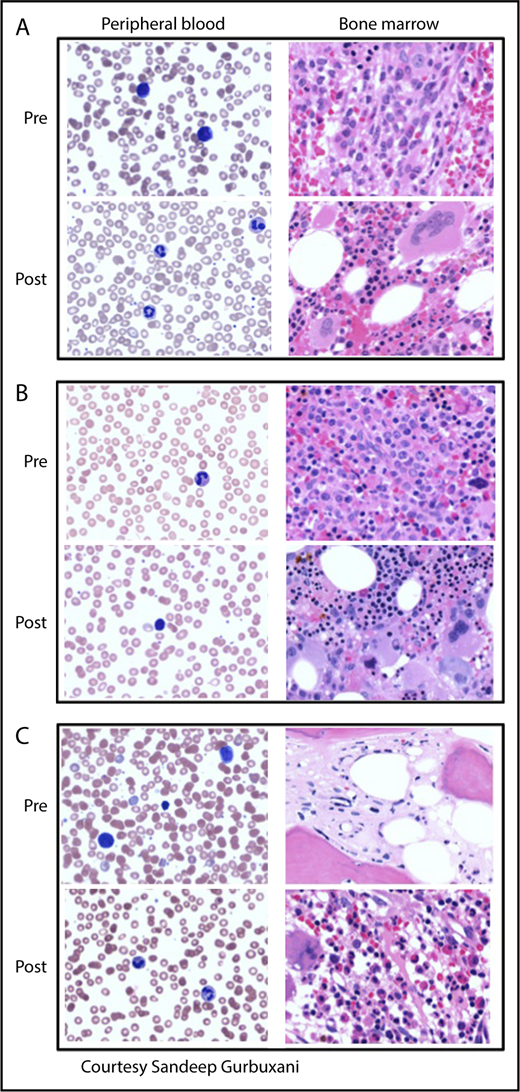
Peripheral blood smears and bone marrow biopsies pre- and posttherapy of MPN-BP. (A) IDH2-mutated MPN-BP. Pretherapy with enasidenib: peripheral smear shows pancytopenia with circulating blasts. Bone marrow is markedly hypercellular and composed largely of blasts intermingled with increased atypical/dyspoietic megakaryocytes. Posttherapy (6 months of therapy with enasidenib): peripheral smear showing normal neutrophils and decline in circulating blasts. Bone marrow is hypercellular without significant increase in blasts but with increased granulopoiesis and atypical megakaryocytes, consistent with the patient’s underlying MPN. (B) TP53-mutated MPN-BP arising in a patient with PV. Pretherapy with decitabine: peripheral smear shows mild thrombocytopenia. Bone marrow is markedly hypercellular with panmyelosis and sheets of blasts. Posttherapy (2 months of therapy with decitabine): peripheral blood demonstrates leukopenia and anemia without circulating blasts. Bone marrow is hypercellular without overt increase in blasts, but with marked proliferation of erythroid precursors as well as megakaryocytes with clustering and atypia, consistent with involvement with the patient’s underlying MPN. (C) MPN-BP arising in a patient with prior postessential thrombocythemia MF. Pretherapy with decitabine: peripheral blood smear demonstrates circulating blasts (24% on differential count), red blood cell anisopoikilocytosis, marked platelet anisocytosis, and hypogranular platelets. Bone marrow biopsy specimen shows mainly fibrosis, with few hematopoietic elements distorted by the markedly increased fibrosis and few irregularly distributed mononuclear cells (blasts), which are difficult to appreciate morphologically. Postallogeneic SCT (day 180): peripheral smear demonstrates normocytic anemia, mild thrombocytopenia, and no circulating blasts. The marrow is hypercellular, with megakaryocytic and granulocytic proliferation and no increase in blasts. Fibrosis is mild at this juncture. Magnification for all images is ×500. Peripheral blood smears are stained with Giemsa; bone marrow biopsy specimens are stained with hematoxylin and eosin.
Peripheral blood smears and bone marrow biopsies pre- and posttherapy of MPN-BP. (A) IDH2-mutated MPN-BP. Pretherapy with enasidenib: peripheral smear shows pancytopenia with circulating blasts. Bone marrow is markedly hypercellular and composed largely of blasts intermingled with increased atypical/dyspoietic megakaryocytes. Posttherapy (6 months of therapy with enasidenib): peripheral smear showing normal neutrophils and decline in circulating blasts. Bone marrow is hypercellular without significant increase in blasts but with increased granulopoiesis and atypical megakaryocytes, consistent with the patient’s underlying MPN. (B) TP53-mutated MPN-BP arising in a patient with PV. Pretherapy with decitabine: peripheral smear shows mild thrombocytopenia. Bone marrow is markedly hypercellular with panmyelosis and sheets of blasts. Posttherapy (2 months of therapy with decitabine): peripheral blood demonstrates leukopenia and anemia without circulating blasts. Bone marrow is hypercellular without overt increase in blasts, but with marked proliferation of erythroid precursors as well as megakaryocytes with clustering and atypia, consistent with involvement with the patient’s underlying MPN. (C) MPN-BP arising in a patient with prior postessential thrombocythemia MF. Pretherapy with decitabine: peripheral blood smear demonstrates circulating blasts (24% on differential count), red blood cell anisopoikilocytosis, marked platelet anisocytosis, and hypogranular platelets. Bone marrow biopsy specimen shows mainly fibrosis, with few hematopoietic elements distorted by the markedly increased fibrosis and few irregularly distributed mononuclear cells (blasts), which are difficult to appreciate morphologically. Postallogeneic SCT (day 180): peripheral smear demonstrates normocytic anemia, mild thrombocytopenia, and no circulating blasts. The marrow is hypercellular, with megakaryocytic and granulocytic proliferation and no increase in blasts. Fibrosis is mild at this juncture. Magnification for all images is ×500. Peripheral blood smears are stained with Giemsa; bone marrow biopsy specimens are stained with hematoxylin and eosin.
HMA therapy in MPN-BP
Therapy with a hypomethylating agent (HMA) was recommended in this case. TP53-mutant AML is relatively chemotherapy resistant.61,62 This issue, coupled with the patient’s older age and poor performance status, made an intensive chemotherapy approach a less attractive option.
Studies evaluating clonal evolution and clonal architecture in MPNs have revealed that TP53 mutations or abnormalities of the TP53 pathway are common in leukemic blasts in MPN-BP.29,30,42 It has been demonstrated that in some cases, TP53 mutations may be present at a low variant allele frequency in the chronic phase of MPNs, and with loss of heterozygosity, there is rapid expansion of the mutant clone associated with transformation to acute leukemia.30
There is an accumulating body of evidence supporting the use of HMAs decitabine and azacitidine in advanced MF, including Ph− MPN-AP or Ph− MPN-BP.63 Most are summaries of retrospective experiences, and only a few reports are focused particularly on MPN-AP/MPN-BP64,,-67 (Table 2). Myelosuppression is the dose-limiting toxicity with the use of these agents, and there is a relative paucity of nonhematologic adverse effects. There are no prospective randomized studies comparing HMA-based approaches with intensive chemotherapy in MPN-BP. Most retrospective series (Table 2) have not shown any significant survival advantage in favor of HMA use compared with intensive chemotherapy. Limitations to these studies include the relatively small sample sizes, heterogeneity of the disease features, and nonrandomized nature of the study designs. The relative tolerability profile of HMA therapy, however, has made this approach an increasingly attractive option, particularly in older patients with borderline or poor performance status or with molecular or cytogenetic risk profiles that would predict chemotherapy resistance.
Alternative schedules (10-day regimens) of azanucleoside therapy in high-risk myeloid neoplasms, including myelodysplastic syndromes and AML, have been explored.68,-70 In a single-arm phase 2 trial in which older adults with previously untreated AML were treated with a 10-day regimen of decitabine in the frontline setting, the CR/CRi rate was 64%, including a CR rate of 50% in patients with complex karyotypes.68 A recent intriguing report incorporating enhanced exome sequencing of samples from individuals with AML or myelodysplastic syndromes treated with the 10-day decitabine regimen revealed that clinical responses were highly correlated with the presence of TP53 mutations in the founding clone.71 The median survival was 12.7 months in the TP53-mutant cohort, and undergoing allogeneic SCT for consolidation had the largest impact on survival.
The patient received decitabine administered at 20 mg/m2 per day on a 10-day schedule, with cycles repeated every 28 days. His second cycle of therapy was complicated by culture-negative febrile neutropenia.
His most recent bone marrow biopsy after 2 cycles of decitabine revealed a 30% cellular bone marrow without an overt increase in blasts but with marked proliferation of erythroid precursors, as well as megakaryocytes with clustering and atypia, reminiscent of the patient’s underlying MPN (Figure 2). The cytogenetic analysis revealed reversion to a normal male karyotype. Fluorescence in situ hybridization analysis (400 cells) was negative for deletion or loss of chromosomes 5 and 7 and was disomy for chromosome 8. NGS analysis revealed JAK2 V617F as the sole pathogenic variant. The mutations in TP53 were no longer detectable (assay sensitivity, 10% mutant alleles). Serum LDH was within normal limits at 214 U/L.
Status
The patient has just completed his fifth cycle of decitabine. Given the myelosuppressive effect of the 10-day schedule and the fact that his bone marrow no longer shows an excess of blasts, he is currently receiving decitabine on a 5-day (maintenance) schedule. His performance status has improved considerably. CBC at this point is significant only for mild anemia, with hemoglobin of 11 g/dL. He has been HLA typed and has several potential matched unrelated donors (MUDs) in the National Bone Marrow Donor Registry. He has recently been referred for a transplantation consultation to evaluate candidacy for allogeneic SCT.
Patient 3
A 60-year-old woman was diagnosed with ET 23 years ago. At that time, she presented with a WBC count of 8 × 109/L, hemoglobin of 128 g/L, and platelet count of 900 × 109/L. Thirteen years after the original diagnosis of ET, she developed splenomegaly (the spleen was palpable at 4 cm below the costal margin) on physical examination. CBC at the time was notable for WBC count of 12.9 × 109/L with a left shift, 3% circulating blasts, hemoglobin of 118 g/L, and platelet count of 382 × 109/L. She had a leucoerythroblastic picture on the peripheral blood smear and teardrop red cells. Her bone marrow biopsy was hypercellular, with moderate reticulin fibrosis and osteosclerosis, associated with increased numbers of atypical megakaryocytes, granulocytic proliferation, and prominent intrasinusoidal hematopoiesis consistent with evolution to post-ET MF. There was no increase in bone marrow blasts, and the bone marrow cytogenetic analysis at the time revealed a normal karyotype. Five years later, she presented with complaints of abdominal discomfort, night sweats, pruritus, and weight loss. There was no history of cytoreductive therapy. The spleen was noted to be massively enlarged, 23 cm below the costal margin and extending into the pelvis. CBC revealed WBC count of 14 × 109/L, hemoglobin of 950 g/L, and platelet count of 331 × 109/L. Serum LDH was significantly elevated at 2130 IU/L. The peripheral blood smear showed leukocytosis with dyspoietic cells and 24% circulating blasts (Figure 2). The marrow biopsy was fibrotic (Figure 2), with few hematopoietic cells. Immunohistochemistry showed focal clusters of CD34+ blasts; the overall blast percentage was difficult to estimate. The bone marrow cytogenetic analysis was uninformative, given the marked fibrosis. The cytogenetic analysis of the peripheral blood revealed a tetraploid clone: 92, XXX[4]/46,XX[16]. An institutional NGS panel was not yet available at that time. Molecular studies performed on the peripheral blood included evaluation for mutations in JAK2, NPM1, FLT3, and CEBPA, all of which were negative. Reverse transcription polymerase chain reaction for BCR/ABL was also negative.
Given the presence of constitutional symptoms and massive splenomegaly, the patient inquired about treatment with the JAK inhibitor ruxolitinib. The potential for ruxolitinib to cause amelioration of symptomatic splenomegaly and constitutional symptoms in advanced MF is well established, and the agent is US Food and Drug Administration approved in MF based on its significant activity in this context.72,-74 The potential utility of ruxolitinib in improving performance status before proceeding with allogeneic SCT in MF, especially in those with significant debilitating symptoms and massive splenomegaly, has been documented in retrospective series.75,76
The available evidence, however (Table 2), does not support the use of single-agent ruxolitinib as an effective strategy in MPN-BP. In 1 prospective trial of ruxolitinib in relapsed and refractory leukemias, 3 of 18 patients with MPN-BP responded, and responses were relatively slow to occur.77 In a subsequent trial using high doses of the agent, at a dose range of 50 to 200 mg twice daily, only 1 patient responded, and infectious complications were frequent, occurring in more than half of patients treated.78 Investigational approaches evaluating the combination of ruxolitinib and HMAs, including decitabine, in MPN-BP are ongoing,79,80 based in part on preclinical evidence of synergy observed between these agents in a murine model of MPN-BP.42
The patient was treated with low-dose subcutaneous decitabine (0.1 mg/kg per day for 10 days administered on days 1-5 and 8-12, with cycles repeated every 4-6 weeks), according to a regimen investigated in a prospective clinical trial in advanced MF at our institution.81 The second cycle of decitabine was complicated by neutropenic fever and Streptococcal mitis bacteremia. After 3 cycles of decitabine, her splenomegaly improved, with the spleen now being palpable at ∼10 cm below the costal margin. Her performance status improved, and her constitutional symptoms were also considerably improved. CBC after 3 cycles revealed a WBC count of 2.4 × 109/L, hemoglobin of 96 g/L, platelet count of 306 × 109/L, and absolute neutrophil count of 0.9 × 109/L, with 5% circulating blasts. Serum LDH also declined significantly to 376 IU/L. The bone marrow biopsy demonstrated persistent extensive fibrosis; blasts did not seem to be obviously increased, but hematopoietic elements were sparse. At this point, an MUD was available; the patient’s performance status was excellent, and she had no significant comorbidities. By the consensus criteria proposed for the assessment of response in MPN-BP,6 this patient would be judged to have a partial response, defined as a decrease in leukemic burden (>50% reduction in blasts) without complete resolution of peripheral blood or bone marrow blasts and with residual MPN features. A question arose regarding the optimal timing of allogeneic SCT in this situation.
Optimal timing of allogeneic SCT and choice of conditioning regimen in MPN-BP
There is lack of a consensus regarding the depth of response required to be able to successfully proceed to allogeneic SCT after treatment for MPN-BP. Patients who proceeded to transplantation in most of the published retrospective series had achieved a CR or CRi or a return to second chronic phase. As expected, achievement of a response before allogeneic SCT was associated with a trend toward improved outcome.46,-48 A subset of patients with refractory disease, however, may still enjoy long-term survival after allogeneic SCT.46,49 The caveat in interpreting the available reports includes the lack uniformity with regard to the criteria used for response assessment in most series and the relatively small numbers of patients proceeding to transplantation. The same limitations preclude any definitive conclusions regarding the optimal conditioning regimen in this context.82 Both reduced-intensity and myeloablative regimens have been used, and the choice of conditioning regimen has not been predictive of transplantation-related mortality, relapse, or survival in published retrospective series.46,47,49,83,84 Donor source has not been an independent predictor of outcome in most reports, although there has been a trend toward increased transplantation-related mortality in some series in patients receiving transplants from MUDs when compared with matched related donors.46,83 There are few data regarding alternative donor transplantation, although long-term survivors after umbilical cord blood transplantation have been reported.84
The patient proceeded to MUD allogeneic SCT with a fludarabine/busulphan conditioning regimen. Her course was complicated by acute graft-versus-host disease of the liver, which came under relatively quick control with corticosteroid therapy. Serial bone marrow biopsies at days 30 and 100 posttransplantation demonstrated persistent MF, but by day 180, the marrow fibrosis had improved (MF grade 1+ of 3). The serial chimerism analysis initially showed mixed donor chimerism. After the development of acute graft-versus-host disease of the liver, chimerism improved to 100% donor in both unfractionated and T-cell compartments. Her bone marrow biopsy at 12 months morphologically revealed minimal reticulin fibrosis of 0 to 1 and trilineage hematopoiesis. She had continued improvement in her splenomegaly, with the spleen being no longer palpable at ∼18 months after allogeneic SCT. She is currently 6 years out from transplantation and doing well.
Special consideration: lymphoid blast transformation arising from Ph− MPNs
Lymphoid blast transformation of the Ph− MPNs is a rare entity limited to case reports in the literature. In a recent case report and review of the existing literature, 18 such cases were summarized.85 A majority of these cases were B-cell acute lymphoblastic leukemias (ALLs), with T-cell ALL occurring rarely. In some of these cases, the leukemia blasts harbored a JAK2 mutation,85,-87 in keeping with earlier observations of the pluripotency of the JAK2 V617F stem/progenitor cells in Ph− MPNs.88 In other cases, however, a clonally distinct origin of the lymphoblasts was demonstrated.89,90 Outcomes were uniformly fatal, with only 2 patients reported alive in the literature.91,92 In both of these cases, intensive induction with an ALL chemotherapy regimen was employed, followed by transplantation in 1 case, once a morphologic remission of the ALL was achieved. Rare case reports of BCR/ABL+ ALL arising in patients with Ph− MPNs have also been reported.90,93 Complete characterization of the leukemia blasts, including exclusion of a BCR/ABL rearrangement, is important in lymphoid blast transformation of a Ph− MPN, given the significant therapeutic implications. We can surmise from the limited experience available, however, that a majority of cases will be BCR/ABL−. An intensive induction approach, followed by allogeneic SCT once a response is achieved, is a reasonable approach in those patients who are fit enough to undergo such therapy.
Conclusion
My approach to treating MPN-BP is summarized in Figure 3 and accompanying Box 1. I strongly favor prospective clinical investigation wherever possible in these diseases. There is a significant need to improve our ability to more accurately predict those individuals who are at high risk for evolution to MPN-BP and who could benefit from earlier intervention. Currently, such intervention is limited to allogeneic SCT, and the recently proposed Mutation-Enhanced International Prognostic Score System,28 if validated, may be useful in refining transplantation decisions in the chronic phase in PMF. The molecular mechanisms underlying transformation to blast phase are increasingly being understood and will pave the way for more effective treatment approaches, but much remains to be accomplished. We need improved preclinical models that accurately recapitulate the human disease, including the clonal heterogeneity, and that can be used for nonclinical drug evaluation. This should be paired with rapid clinical translation of the most promising agents and combinations. There is currently a paucity of trials in existence (Table 3) that include acute phase MPN/MPN-BP. Given the clinical and molecular heterogeneities of these diseases, subset-specific therapy is predicted to be necessary to improve outcomes. Advances in allogeneic SCT will continue to be important in affording patients with MPN-BP a chance of cure, especially because this modality is increasingly being extended to older adults.94 The potential utility of minimal residual disease monitoring and posttransplantation maintenance approaches requires further investigation. Ultimately, the hope is that a multipronged approach will bring us closer to improving outcomes for high-risk Ph− MPNs, including those evolving to MPN-BP.
My approach to treating MPN-BP:
I favor offering the option for therapy to as many patients as possible; survival is very short when a supportive care–only approach is adopted.
Because allogeneic SCT is the only approach shown to have the potential to provide durable remissions, I prospectively HLA type my patients and start looking into donor options as soon as the diagnosis is made.
I perform NGS analysis on the leukemia blasts at diagnosis to evaluate for potentially actionable mutations. In many patients, particularly those who develop MPN-BP from a background of MF (which is a majority of patients), a peripheral blood sample is generally adequate for performing such an analysis, because the marrow is frequently inaspirable.
I strongly favor enrollment in a clinical trial whenever possible, especially clinical trials designed with a particular focus on this patient population.
I tailor therapy wherever possible if there is an actionable mutation for which there is a targeted therapeutic approach available. For IDH1/2-mutant disease, I favor a clinical trial that would allow access to an IDH1 or IDH2 inhibitor, either as a single agent or in combination with an HMA and, for fit patients, in combination with chemotherapy.
For TP53-mutant disease, I strongly favor clinical trial enrollment. In the absence of a trial, I am inclined to favor HMA therapy over a 7 + 3 approach from a risk/benefit standpoint, especially in older adults who are less fit.
For those patients (with other mutations) who are fit for transplantation and in whom a donor will be readily available, I favor intensive chemotherapy. It is important to stress that remissions are not durable with intensive chemotherapy alone. Therefore, preparations must be made to move forward with allogeneic SCT as soon as remission or significant cytoreduction is achieved. There are no standard guidelines regarding how deep the remission needs to be before SCT.
Many patients will not have a donor immediately available or be transplantation eligible, and some who do may not wish to undergo intensive chemotherapy, given the uncertainty that this will lead to a long-term leukemia-free survival. For those patients, I favor HMA-based therapy in a clinical trial, if 1 is available. In the absence of a clinical trial, I favor the use of decitabine or azacitidine, given the emerging experience in MPN-BP. Although these agents are designed for outpatient use, it is essential to stress that they are particularly myelosuppressive in the MPNs with underlying fibrosis, especially when administered on a 10-day schedule.
I advocate meticulous attention to supportive care, including blood product support and use of prophylactic antimicrobials in the setting of neutropenia. I use growth factor support according to established guidelines in the presence of febrile neutropenia.

How I treat Ph−MPN-BP. My approach to the treatment of MPN-BP is summarized. The accompanying text in Box 1 outlines in greater detail the treatment algorithm illustrated here. *The promise of IDH1/2 inhibitor–based therapy is not yet validated in MPN-BP and is recommended in the context of clinical trial participation. †HMA-based therapy has not been validated as superior to intensive chemotherapy (ICT) in TP53-mutated cases, and the choice of 1 over the other must be based on the individual case.
How I treat Ph−MPN-BP. My approach to the treatment of MPN-BP is summarized. The accompanying text in Box 1 outlines in greater detail the treatment algorithm illustrated here. *The promise of IDH1/2 inhibitor–based therapy is not yet validated in MPN-BP and is recommended in the context of clinical trial participation. †HMA-based therapy has not been validated as superior to intensive chemotherapy (ICT) in TP53-mutated cases, and the choice of 1 over the other must be based on the individual case.
Selected trials that include patients with Ph− MPN-AP or MPN-BP
| Agent | Mechanism of action | Trial phase | Disease | Clinicaltrials.gov ID |
|---|---|---|---|---|
| Ruxolitinib + decitabine | JAK inhibition + HMA | 1/2 | MPN-AP/MPN-BP | NCT02076191 |
| Ruxolitinib + decitabine | JAK inhibition + HMA | 1/2 | MPN-AP/MPN-BP | NCT02257138 |
| SGI-110 | HMA | 2 | Ph− MPN, including MF-AP | NCT03075826 |
| Various | Various molecularly targeted | 1/2 | AML-TN in older adults; includes MPN-BP | NCT03013998 |
| Enasidenib or ivosidenib + azacitdine | IDH2 or IDH1 inhibition + HMA | 1/2 | AML-TN; includes MPN-BP | NCT02677922 |
| Azacitidine ± ivosidenib | HMA ± IDH1 inhibition | 3 | AML-TN; includes MPN-BP; not candidate for IC | NCT03173248 |
| Ivosidenib or enasidenib + cytarabine/daunorubicin | IDH1 or IDH2 inhibition + IC | 1 | AML-TN; includes MPN-BP | NCT02632708 |
| Venetoclax + ivosidenib | BCL2 inhibition + IDH1 inhibition | 1 | Advanced heme malignancies, including MPN-AP/MPN-BP | NCT03471260 |
| Decitabine | HMA | 2 | TP53-mutated R/R AML | NCT03063203 |
| Decitabine + pevonedistat | HMA + NAE inhibition | 1 | R/R AML, including secondary AML; AML-TN, age >60 y; not candidate for IC | NCT03009240 |
| Azacitidine + selumetinib | HMA + MEK inhibition | 1 | Advanced myeloid neoplasms, including MPN-AP | NCT03326310 |
| XmAb14045 | CD123 BiTE antibody | 1 | Advanced CD123+ heme malignancies, including R/R AML | NCT0273012 |
| Agent | Mechanism of action | Trial phase | Disease | Clinicaltrials.gov ID |
|---|---|---|---|---|
| Ruxolitinib + decitabine | JAK inhibition + HMA | 1/2 | MPN-AP/MPN-BP | NCT02076191 |
| Ruxolitinib + decitabine | JAK inhibition + HMA | 1/2 | MPN-AP/MPN-BP | NCT02257138 |
| SGI-110 | HMA | 2 | Ph− MPN, including MF-AP | NCT03075826 |
| Various | Various molecularly targeted | 1/2 | AML-TN in older adults; includes MPN-BP | NCT03013998 |
| Enasidenib or ivosidenib + azacitdine | IDH2 or IDH1 inhibition + HMA | 1/2 | AML-TN; includes MPN-BP | NCT02677922 |
| Azacitidine ± ivosidenib | HMA ± IDH1 inhibition | 3 | AML-TN; includes MPN-BP; not candidate for IC | NCT03173248 |
| Ivosidenib or enasidenib + cytarabine/daunorubicin | IDH1 or IDH2 inhibition + IC | 1 | AML-TN; includes MPN-BP | NCT02632708 |
| Venetoclax + ivosidenib | BCL2 inhibition + IDH1 inhibition | 1 | Advanced heme malignancies, including MPN-AP/MPN-BP | NCT03471260 |
| Decitabine | HMA | 2 | TP53-mutated R/R AML | NCT03063203 |
| Decitabine + pevonedistat | HMA + NAE inhibition | 1 | R/R AML, including secondary AML; AML-TN, age >60 y; not candidate for IC | NCT03009240 |
| Azacitidine + selumetinib | HMA + MEK inhibition | 1 | Advanced myeloid neoplasms, including MPN-AP | NCT03326310 |
| XmAb14045 | CD123 BiTE antibody | 1 | Advanced CD123+ heme malignancies, including R/R AML | NCT0273012 |
BiTE, bispecific T cell engaging; NAE, Nedd8-activating enzyme inhibitor; TN, treatement naïve.
Acknowledgment
The author is indebted to Sandeep Gurbuxani in the Section of Hematopathology, The University of Chicago Medicine, for creating Figure 2 (photomicrographs of the pathologic specimens of cases of MPN-BP).
Authorship
Contribution: O.O. conceived and wrote this article.
Conflict-of-interest disclosure: O.O. has received research funding (paid to her institution) from Agios, AstraZeneca, AbbVie, Celgene, CTI/Baxalta, Gilead, Incyte, Janssen, NS-Pharma, and Oncotherapy Sciences; has served on advisory boards for Jazz Pharma, Pfizer, DavaOncology, CTI BioPharma, Celgene, and AbbVie; has received honoraria from the American Board of Internal Medicine (Medical Oncology Board Exam Committee), American Society of Hematology, American Society of Clinical Oncology, National Institutes of Health Molecular and Cellular Hematology Study Section, and American Society for Clinical Pathology; and has membership on the medical advisory board of the Aplastic Anemia/MDS International Foundation (no financial compensation involved).
Correspondence: Olatoyosi Odenike, University of Chicago Comprehensive Cancer Center, The University of Chicago Medicine, 5841 S Maryland Ave, MC 2115, Chicago, IL 60637; e-mail: todenike@medicine.bsd.uchicago.edu.
