

If hemophagocytic lymphohisticytosis (HLH)–causing mutations are found in a well child, should that patient receive a bone marrow transplant? In this issue of Blood, have collected a unique subset of patients with biallelic mutations causing HLH and provide an important recommendation for the management of asymptomatic carriers (ACs) with the same mutation as affected siblings, index cases (ICs).1
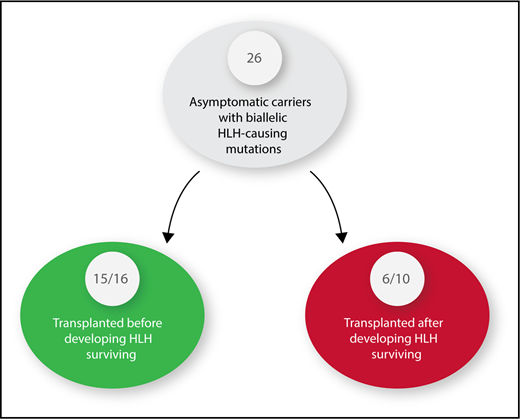
Twenty-six asymptomatic carriers of HLH-causing mutations were identified. Sixteen received hematopoietic stem cell transplants before developing HLH. Ten had to be treated for HLH and were then transplanted.
Twenty-six asymptomatic carriers of HLH-causing mutations were identified. Sixteen received hematopoietic stem cell transplants before developing HLH. Ten had to be treated for HLH and were then transplanted.
HLH patients, whether children or adults, are among the most difficult to diagnose and challenging to treat of any patients cared for by hematology/oncology specialists. The overall survival is 50% to 60% whether patients have an identified HLH-causing mutation or not.2,3 Delay in diagnosis, inadequate response to initial or salvage therapy, infections, and inflammation-related organ damage contribute to this poor outcome. HLH patients with a mutation are best treated with hematopoietic stem cell transplantation (HSCT) after control of the disease is achieved by treatment with decadron and etoposide. In the course of identifying a suitable donor for HSCT, one may find a sibling who is an AC with the same biallelic mutation responsible for HLH in the IC. Until the report by Lucchini et al, clinicians had no data to inform them as to whether it is better to perform an HSCT before or after the AC has developed HLH. Although transplant-related mortality (TRM) has been markedly improved with reduced intensity conditioning, it is still very significant, and convincing parents and insurance companies to proceed with a transplant in a well child is an ethical dilemma.4 The most striking fact of this article is that 15 of 16 ACs transplanted before development of HLH survived compared with only 6 of 10 transplanted after development of HLH, a statistically significant difference even with the small numbers of patients (see figure). The authors emphasize that patients with perforin mutations are most likely to activate early in life, as has been reported by others,5,6 thus making ACs with this mutation ideal candidates for early HSCT. Those with the MUNC 18-2 mutation who are known to present at a later age and have a milder form of HLH, probably because they can recover natural killer cell activity after chemotherapy and may be observed to see if a HSCT is truly needed.7 Only 1 of 5 ACs with MUNC 18-2 deficiency developed HLH, with 2/5 currently being almost 4 years without any evidence of HLH. MUNC 18-2 deficiency, although the second most common HLH mutation, is one with the most complex clinical phenotypes because ectopic expression of wild-type STXBP2 could overcome the MUNC 18-2 deficiency.8 The ectopic STXBP2 expression could lead to increased variability of organs involved, making it very difficult to predict outcomes. HSCT is the only curative option for HLH patients with HLH-associated gene mutations, nonresponse to primary therapy, or central nervous system involvement, but most patients will experienced major treatment-related toxicities when transplanted after active HLH.4 It is important to note that there were no TRMs in the 16 ACs who were transplanted prior to the development of HLH making this approach even more attractive.
A preemptive HSCT approach is currently favored in many other congenital immune deficiencies with encouraging improvement in overall outcomes.4 Lucchini et al successfully showed the marked improved survival of early HSCT for patients with HLH while asymptomatic. Siblings of HLH patients should be screened for the specific mutation found in an IC. If a perforin mutation is found, then the recommendation is to act while asymptomatic to more likely achieve a cure, but if any other mutations with milder phenotypes are found, then it may be reasonable to wait and watch. Hopefully, Lucchini et al will continue collecting cases and other centers will collaborate with them so a more robust number of patients can be reported as a validation of this preliminary result.
Conflict-of interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal