

In this issue of Blood, by comparing wild-type vs erythroid-specific ferroportin knockout mice, 1 have shown that ferroportin-mediated export of iron from erythroblasts and erythrocytes contributes to the systemic iron economy and prevents hemolysis.
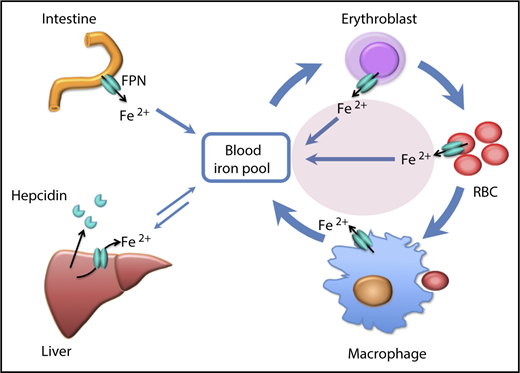
Illustration of the contribution (pink oval) of iron efflux from erythroid cells to the iron economy of the body. FPN, ferroportin; RBC, red blood cell. The figure has been modified from Figure 6 in the article by Zheng et al that begins on page 2078.
Illustration of the contribution (pink oval) of iron efflux from erythroid cells to the iron economy of the body. FPN, ferroportin; RBC, red blood cell. The figure has been modified from Figure 6 in the article by Zheng et al that begins on page 2078.
“When a dog bites a man, that is not news, because it happens so often. But if a man bites a dog, that is news” (attributed to Alfred Harmsworth [1865–1922], a British newspaper magnate). The release of iron by erythroid cells during their normal life span is certainly “a man bites dog” story. Why would erythroblasts and erythrocytes give up iron they had accumulated at such a great metabolic cost?
Humans evolved under conditions of endemic iron deficiency, and this remains the most common nutrient deficiency and the main cause of anemia worldwide. In the adult human, erythrocyte hemoglobin contains most of the iron in the body (2-2.5 g of 3-4 g total) and the erythroblasts in the marrow are voracious consumers of iron (about 90% of ∼25 mg/d iron moving through the circulating plasma transferrin compartment). However, all metabolically active cells require iron, and the central nervous system, skeletal muscle, and heart are known to be particularly sensitive to iron deprivation.2-4 Misallocation of scarce iron to erythroid cells at the expense of other tissues would have grave consequences for organismal function. Accordingly, erythrocyte production is closely regulated by the concentration of transferrin-bound plasma iron and by the concentration of iron in erythroblasts, causing erythropoiesis to slow down during periods of iron deficiency, and so leaving more iron for other tissues. At the other extreme of iron availability, some cells and tissues can be damaged by the excessive cellular accumulation of chemically reactive forms of iron, causing injury, fibrosis, and carcinogenesis.5
The major sources of plasma iron include absorbed dietary iron from the duodenum, iron recycled by macrophages from senescent erythrocytes, and iron retrieved from storage in hepatocytes (see figure). Iron flows from these cells into plasma through the sole known cellular iron exporter ferroportin,6 under control of its ligand hepcidin, a peptide hormone secreted by hepatocytes.7,8 Building on a previous publication on the protective role of erythroid ferroportin-mediated iron efflux in malaria,9 Zhang et al now present evidence that during their normal life span, erythroblasts and erythrocytes release iron that importantly contributes to systemic iron economy (see figure).
The earliest clue to the potential ability of erythroid cells to release iron to plasma was their abundant expression of an iron-regulatory element (IRE)–deficient isoform of ferroportin messenger RNA (mRNA), also found in duodenal enterocytes.10 In both tissues, the lack of the 5′IRE in ferroportin mRNA implies that translation of this isoform into ferroportin protein would not be repressed by cellular iron deficiency, potentially allowing the duodenum and erythroblasts to release iron during systemic iron shortage, despite their own iron-deficient status. Because most of the iron in the body is in erythroblasts and erythrocytes, the release of a small fraction of this iron could provide sufficient amounts of this essential nutrient to organs sensitive to iron deprivation,2-4 without excessive iron depletion of the erythroid compartment. The contribution of this source of iron to the systemic iron economy is now demonstrated by Zhang et al in mice that were genetically engineered to lack erythroid ferroportin. Surprisingly, the resulting loss of iron export from erythroblasts and erythrocytes led to ∼20% decrease in serum iron concentration and transferrin saturation. This represents an underestimate of the contribution of iron efflux from live erythroid cells because, as discussed below, the ferroportin-deficient erythrocytes have a shorter life span, thus terminally returning all their iron to the organism at a higher rate.
During their development in the marrow, erythroblasts take up nearly all available iron to rapidly synthesize enormous amounts of heme and hemoglobin. Here ferroportin may function to prevent iron-mediated damage to erythroblasts by releasing excess iron not used for hemoglobin synthesis. Mature human erythrocytes, one quarter of all the cells in the body, then spend 120 days in blood circulation, subjecting their extraordinarily high concentration of iron-rich hemoglobin to almost 200 000 cycles of redox stress, whereas in mice, erythrocytes experience >500 000 such cycles during 40 days in blood circulation. Here ferroportin is thought to remove iron leaking from damaged hemoglobin of circulating erythrocytes. Consistent with the proposed mechanism, mice lacking erythroid ferroportin had mild chronic hemolytic anemia as well as increased sensitivity to hemolytic stress. How iron is released from the heme of hemoglobin and how it reaches ferroportin remain to be determined. Interestingly, in a different condition (ie, iron deficiency), iron surplus in ferroportin-deficient erythroid cells enhanced hemoglobin synthesis even during iron deficiency, resulting in larger erythrocytes that those from iron-deficient wild-type mice.
Erythroid cells represent an extreme of cell biology that appears to require counterintuitive adaptions, of which their ability to release iron during their life span is only the latest example.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal