

Historically, detailed studies of rare congenital platelet defects have provided unique insights into key proteins involved in human platelet function. In this issue of Blood, use advanced genomic sequencing/functional analysis to identify a novel role for ephrin transmembrane receptor subclass B2 (EPHB2) in regulating platelet activation.1
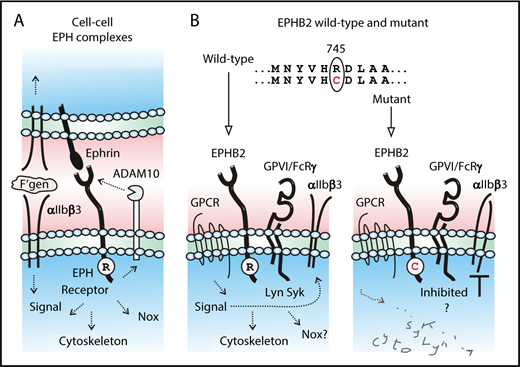
(A) At cell-cell boundaries, ephrin receptor/ephrin ligand complexes regulate cellular adhesion and signaling, including ligand-induced outside-in signaling of αIIbβ3. (B) Compared with wild-type, a newly identified Arg745Cys (R745C) mutation in the tyrosine kinase domain of EPHB2 is associated with impaired platelet activation in response to agonists acting at G protein-coupled receptors (GPCRs) or glycoprotein VI (GPVI) and attenuated phosphorylation of Lyn, Syk, and FcRγ and inside-out activation of αIIbβ3. See the text for details and references.
(A) At cell-cell boundaries, ephrin receptor/ephrin ligand complexes regulate cellular adhesion and signaling, including ligand-induced outside-in signaling of αIIbβ3. (B) Compared with wild-type, a newly identified Arg745Cys (R745C) mutation in the tyrosine kinase domain of EPHB2 is associated with impaired platelet activation in response to agonists acting at G protein-coupled receptors (GPCRs) or glycoprotein VI (GPVI) and attenuated phosphorylation of Lyn, Syk, and FcRγ and inside-out activation of αIIbβ3. See the text for details and references.
The family of ephrin transmembrane tyrosine kinase receptors (subclasses A and B) and their membrane-associated ephrin ligands are highly conserved across multicellular organisms, from primitive invertebrates to mammals, consistent with their crucial role in regulating cell-cell interactions.2 The multiple phosphatidylinositol-linked (subclass A) or transmembrane (B) ephrin ligands form transcellular complexes with their specific ephrin receptors. Ephrin receptors consist of a conserved extracellular ephrin ligand-binding domain, cysteine-rich and fibronectin-like domains, a transmembrane domain, and an intracellular tyrosine kinase domain. These ephrin/receptor complexes are expressed within cell-cell boundaries and regulate cell signaling pathways, rearrangement of the actin-containing cytoskeleton, redox regulation through activation of nicotinamide adenine dinucleotide phosphate oxidase (Nox), and activation of proteases, including a disintegrin and a metalloproteinase-10 (ADAM10) involved in downregulating EPH receptor expression.3 Platelets express the Eph kinases, EphA4 and EphB1, and their ligand, ephrinB1, and contribute to thrombus stability.4 More recently, platelets have also been found to express EPHB2.5 Platelets from genetically modified mice in which the entire EPHB2 cytoplasmic tail was replaced by β-galactosidase demonstrated not only a role for ephrin/receptor complexes in the gap between adhesive platelets, in particular regulating platelet integrin αIIbβ3 outside-in signaling in thrombus formation and clot retraction, but also a contact-independent role for EPHB2 in regulating thrombin and collagen signaling pathways and αIIbβ3 inside-out signaling.5 In this issue, Berrou et al have confirmed a role for EPHB2 in regulating these platelet activation pathways independently of receptor ligation (see figure).
Two patients, a brother and a sister of consanguineous parents, were initially investigated in their early teens. They exhibited spontaneous subcutaneous bleeding and severe bleeding after minor wounds.1 In the brother, there was also childhood anemia because of excessive gastrointestinal bleeding. Neither parent showed any bleeding tendency. The platelet counts for all family members were within the normal range or moderately reduced. Patient platelet aggregation was compromised in response to adenosine 5′-diphosphate (ADP), arachidonic acid, and collagen, but not to ristocetin, while dense granule number and secretion appeared normal.1 Whereas in preceding decades identifying specific causative factors or mutations associated with unexplained bleeding was an arduous endeavor,6 a key feature of the present study was application of whole exome sequencing to identify 6 nonsynonymous homozygous rare variants shared by the 2 patients, which together with other evidence described in detail in the publication enabled a homozygous 2233C>T (Arg745Cys) missense mutation in the gene for EPHB2 receptor tyrosine kinase to be identified as the most likely underlying cause of the defect.1 Both parents were heterozygous for this mutation, which is located within the highly conserved intracellular tyrosine kinase motif immediately before Asp746, the presumed proton acceptor essential for the adenosine triphosphate–dependent kinase active site of EPHB2. This disruption of tyrosine kinase activity by the Arg745Cys mutation was confirmed by diminished convulxin-dependent tyrosine phosphorylation at Tyr594/Tyr604 despite similar EPHB2 expression levels in the patient and control platelets.
Whereas previous research involving the EPHB1/B2 complex indicated its significant role in regulating integrin αIIbβ3 outside-in signaling and function,4,5 αIIbβ3 outside-in signaling (clot retraction, platelet adhesion, and spreading on fibrinogen) was only modestly affected in the patient platelets. Instead the patient platelets showed defective activation of αIIbβ3 (inside-out signaling) in response to activation by agonists acting at GPCRs (ADP, thrombin-receptor ligands) or agonists acting at GPVI (collagen, the snake toxin convulxin) and impaired thrombus formation on a collagen-coated surface under flow ex vivo at an arterial-equivalent shear rate of 1500/s. Tyrosine phosphorylation of the signaling molecules, Lyn kinase, spleen tyrosine kinase (Syk), and Fc receptor γ-chain (FcRγ), all previously implicated in GPVI/FcRγ complex signaling, was markedly reduced in the absence of platelet-platelet contact suggesting a direct or indirect role for EPHB2 in regulating GPVI-dependent platelet activation (see figure panel B). Convulxin acting at GPVI induced Tyr594/Tyr604 phosphorylation of wild-type but not mutant EPHB2. These findings were confirmed in overexpression studies of wild-type or mutant EPHB2 in rat basophilic leukemia (RBL-2H3) cells (that express GPVI/FcRγ complex), where the Arg745Cys mutation inhibited convulxin-dependent EPHB2 phosphorylation, but not ephrin ligand-induced EPHB2 clustering, the former supporting the relevance of Arg745/Asp746 in disrupting EPHB2 tyrosine phosphorylation.
These findings raise interesting questions regarding the potential mechanism for how mutant EPHB2 acts to inhibit GPVI/FcRγ- and/or GPCR-dependent signaling pathways in platelets, apparently without the requirement for platelet-platelet contact/ephrin ligand engagement, and despite no demonstrable differences in expression levels of these other receptors. It is most likely to be a combination of factors contributing to the platelet defect. Notably, electron microscopy revealed significant differences in shape and fragmentation propensity in Arg745Cys EPHB2 platelets, consistent with a role for EPHB2 in megakaryocytes/circulating platelets in platelet production and cytoskeletal maintenance. Lack of phosphorylation of mutated EPHB2 could also impact upon localization or activity of recruited signaling-pathway kinases and/or phosphatases, affecting signaling by GPVI. Interestingly, like ephrin/ephrin receptor complexes,3,4 engagement of GPVI leads to activation of both Nox1/2 and ADAM10,7-9 involved respectively in generation of intracellular reactive oxygen species via Syk-dependent and Syk-independent pathways, and in ectodomain metalloproteinase-mediated shedding. In this regard, the cytoplasmic domain of human platelet GPVI also contains an unpaired thiol, leading to rapid GPVI disulfide-dependent dimerization following agonist stimulation,10 and although there is no evidence as yet, it is intriguing to speculate on aberrant interactions involving Cys745 in mutant EPHB2, particularly if EPHB2 was normally associated with the GPVI/FcRγ complex.
In summary, the identification of a specific mutation in EPHB2 in 2 family members with severe bleeding defects and defective GPVI/GPCR-dependent signaling pathways provides a new understanding of human platelet production and function. It will be interesting in future to identify and characterize this and other mutations in populations that help define precise mechanisms that regulate platelet reactivity.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
