

Key Points
CrbnI391V mice degrade known thalidomide derivative targets and recapitulate thalidomide-induced cytopenias and teratogenicity.
Degradation of Ck1α is sufficient to explain the in vivo therapeutic window of lenalidomide in del(5q) myelodysplastic syndrome.

Abstract
Thalidomide and its derivatives, lenalidomide and pomalidomide, are clinically effective treatments for multiple myeloma and myelodysplastic syndrome with del(5q). These molecules lack activity in murine models, limiting investigation of their therapeutic activity or toxicity in vivo. Here, we report the development of a mouse model that is sensitive to thalidomide derivatives because of a single amino acid change in the direct target of thalidomide derivatives, cereblon (Crbn). In human cells, thalidomide and its analogs bind CRBN and recruit protein targets to the CRL4CRBN E3 ubiquitin ligase, resulting in their ubiquitination and subsequent degradation by the proteasome. We show that mice with a single I391V amino acid change in Crbn exhibit thalidomide-induced degradation of drug targets previously identified in human cells, including Ikaros (Ikzf1), Aiolos (Ikzf3), Zfp91, and casein kinase 1a1 (Ck1α), both in vitro and in vivo. We use the CrbnI391V model to demonstrate that the in vivo therapeutic activity of lenalidomide in del(5q) myelodysplastic syndrome can be explained by heterozygous expression of Ck1α in del(5q) cells. We found that lenalidomide acts on hematopoietic stem cells with heterozygous expression of Ck1α and inactivation of Trp53 causes lenalidomide resistance. We further demonstrate that CrbnI391V is sufficient to confer thalidomide-induced fetal loss in mice, capturing a major toxicity of this class of drugs. Further study of the CrbnI391V model will provide valuable insights into the in vivo efficacy and toxicity of this class of drugs.
Introduction
The use of thalidomide as a treatment for morning sickness in the 1950s resulted in the birth of more than 10 000 children with phocomelia and other characteristic malformations, as well as an unknown but likely high number of miscarriages.1,2 Since that time, thalidomide and its derivatives, lenalidomide and pomalidomide, have demonstrated clinical efficacy in B-cell malignancies, including multiple myeloma,3-7 mantle cell lymphoma,8 and chronic lymphocytic leukemia.9,10 In addition, lenalidomide has striking activity in myelodysplastic syndrome (MDS) with somatic deletion of the long arm of one copy of chromosome 5 (del(5q)), and complete cytogenetic remission is seen in ∼50% of del(5q) MDS patients.11-14 Despite our rapidly advancing understanding of the molecular mechanism of these drugs, in vivo studies of thalidomide derivatives have been limited by their lack of activity in rodent models.15,16
Thalidomide derivatives alter the substrate specificity of the CRL4CRBN E3 ubiquitin ligase.17-24 These drugs bind to cereblon (CRBN),25 the substrate adaptor of an E3 ligase complex comprised of CUL4A, ROC1, and DDB1. Drug binding induces the recruitment of specific protein substrates to CRL4CRBN, resulting in their ubiquitination and degradation by the proteasome.17-24 Drug-induced degradation of 2 lymphoid transcription factors, Ikaros (IKZF1) and Aiolos (IKZF3), provides a mechanistic basis for the efficacy of thalidomide, lenalidomide, and pomalidomide in the treatment of B-cell malignancies such as multiple myeloma.18,19,22 Casein kinase 1α (CK1α, encoded by the CSNK1A1 gene) is also targeted for lenalidomide-induced degradation.17 CSNK1A1 is located within the minimally deleted region for MDS with del(5q) and is expressed at haploinsufficient levels in neoplastic cells.14,26 Degradation of CK1α could thus provide a mechanistic basis for lenalidomide’s therapeutic window in del(5q) MDS, although it has not been possible to test this hypothesis in vivo because of the absence of a murine model with drug activity.
Ikzf1, Ikzf3, and Ck1α are not degraded in mouse cells treated with thalidomide analogs.17 We previously determined that mouse Crbn with substitution of Ile391 with the corresponding human valine residue is capable of mediating drug-induced degradation of Ikzf1, Ikzf3, and Ck1α in vitro.17 Both mouse and human CRBN bind thalidomide derivatives,25 but Ile391 in murine Crbn causes steric hindrance that prevents recruitment of these substrates.17,23 Here, we report the development of a CrbnI391V knock-in mouse model in which a single amino acid in mouse Crbn is replaced with the corresponding human residue, rendering the animals responsive to thalidomide and its derivatives. The CrbnI391V model provides the first opportunity to study thalidomide derivatives in mice, for which many experimental and genetic models are available. By combining CrbnI391V with a previously characterized Csnk1a1 knockout allele, we demonstrate that haploinsufficiency for Ck1α is sufficient to explain lenalidomide’s in vivo therapeutic window in del(5q) MDS.
Methods
Animal studies
CrbnI391V mice were generated via homologous recombination by Ingenious Targeting Labs (Ronkonkoma, NY) on a C57BL/6 background. The FRT sites and Neomycin cassette were removed by crossing with C57BL/6 FLP mice.
Conditional knockout mice for Csnk1a1 (B6.129(Cg)-Csnk1a1 <tm1.1Ybn>/J) and Rps14 (B6.129(Cg)-RPS14 <tm1.1Ybn>/J) have been previously described.27,28 Mx1-Cre mice (Tg(Mx1-cre)1Cgn, stock number 002527) and Trp53 conditional knockout mice (B6.129S2-Trp53 <tm1Tyj>/J, stock number 002101) were obtained from The Jackson Laboratory (Bar Harbor, ME). Excision of conditional alleles was induced with 3 intraperitoneal injections of 0.2 mg high-molecular-weight poly(I:C) (Invivogen). CrbnI391V mice will be sent to the Jackson Laboratory for distribution.
In vivo treatment
Lenalidomide, thalidomide, and pomalidomide were obtained from Ark Pharm (Arlington Heights, IL). Stock solutions of drugs were prepared in dimethyl sulfoxide (DMSO), stored at −80°C, and diluted to 10% to 20% DMSO with sterile saline directly before administration. Drugs were dosed by gavage using Instech Laboratory’s (Plymouth Meeting, PA) flexible feeding tubes. Mice were randomized to treatment groups using the rand() function of Excel (Microsoft).
Teratogenicity experiments
Females were checked each morning for the presence of a mating plug and were removed from the mating cage when a plug was found. Noon on that day was specified as e0.5. Dams who lost weight from e0.5 to e4.5 were excluded from randomization and maternal day e4.5 weights were used to calculate treatment doses. When indicated, dams were euthanized at e10.5 or e16.5 to determine the number of viable pups. Otherwise, the number and phenotype of the pups was determined the day after birth and at weaning.
In vivo transplant
Whole bone marrow was obtained from the long bones and spine of donor mice by crushing, filtration, and erythrocyte lysis in BD Pharm Lyse. Recipients were irradiated with 2 doses of 475 cGy at least 4 hours apart. Immediately following the second dose of irradiation, 5 to 10 million donor whole bone marrow cells/recipient were administered in Hanks buffered salt solution by retro-orbital injection. Excision of floxed alleles was induced with poly(I:C) at least 5 weeks after transplant. Treatment with thalidomide derivatives was initiated at least 5 weeks after poly(I:C) treatment.
Generation of Hoxb8 cell lines and Hoxb8 cell line experiments
Immortalized myeloid progenitor cell lines were generated via estrogen-regulated expression of Hoxb8 in c-Kit+ bone marrow cells as previously described.29
Western blots
Western blots were performed as previously described.17,18 Antibodies were: anti-Ck1α (C-19, Santa Cruz Biotechnology), anti-Ikzf1 (H-100, Santa Cruz Biotechnology), anti-IKZF3 (Imgenex), anti-Zfp91 (A303-245A, Bethyl Laboratories), anti-Actin HRP (ab20272, abcam), goat anti-rabbit horseradish peroxidase (HRP, Prometheus Labs), and bovine anti-goat HRP (Jackson Immunolabs).
Isolation of bone marrow and c-Kit+ cells
A single-cell suspension of bone marrow was obtained by crushing long bones and spine with a mortar and pestle. Resuspension in BD Pharm Lyse was used to lyse erythrocytes, except for Ter119/CD71 staining. C-Kit+ cells were isolated with CD117 microbeads (Miltneyi) and an AutoMACS Pro (Miltenyi) and grown in Serum-Free Expansion Media (StemCell Technologies) supplemented with 50 ng/mL mTPO (Peprotech) and 50 ng/mL mSCF (Peprotech).
Isolation of T cells
A single-cell suspension of mouse splenocytes was obtained by crushing spleens through a 100-µm cell strainer. Erythrocytes were lysed by resuspension in BD Pharm Lyse. T cells were isolated with a Pan T Cell Kit II or CD3 Microbeads and an AutoMACS Pro (all Miltenyi).
Isolation of peripheral blood and blood counts
Peripheral blood was obtained via retroorbital bleeds. Blood counts were obtained from a Hemavet (Drew Scientific) or ADVIA (Siemens Heathcare) using mouse settings. The percentage of B220+, CD3+, and CD11b+ Gr1+ cells in erythrocyte-lysed peripheral blood was determined by flow cytometry and was multiplied by the white blood cell count to calculate absolute concentrations.
Proteomics
Tandem mass tag (TMT) proteomics was conducted according to standard protocols, as detailed in the supplemental Methods (available on the Blood Web site).
Flow cytometry
Flow cytometry was performed on an LSRII or Canto II (BD). Specific antibodies are listed in the supplemental Methods.
ELISA
T cells were harvested, stimulated with Dynabead Mouse T cell activator, and treated with lenalidomide, pomalidomide, or DMSO. After 48 hours, interleukin-2 (IL-2) in the supernatant was measured with a mouse IL-2 Quantikine enzyme-linked immunosorbent assay (ELISA) Kit (R&D Technologies).
Alignment of CRBN sequences
Protein sequences of CRBN homologs were obtained from UniProt and aligned with Clustal Ω (1.2.4). Supplemental Table 3 lists the UniProt identifiers and residues included in the alignment.
Results
Thalidomide derivatives induce degradation of substrates in CrbnI391V knock-in mice and increase IL-2 production by T cells
To enable the therapeutic activity and toxicity of thalidomide derivatives in vivo, we generated a knock-in mouse model in which Ile391 in murine Crbn was substituted with valine by homologous recombination (supplemental Figure 1A). Integration of the targeting vector was assessed by Southern blot and polymerase chain reaction at the Crbn locus (supplemental Figure 1B-C). Pups homozygous for the I391V mutation were born at the expected frequency (supplemental Figure 1D). The I391V amino acid change was confirmed in peripheral blood by DNA sequencing (supplemental Figure 1E) and did not impair the expression of Crbn in hematopoietic cells (supplemental Figure 1F).
We first assessed whether cells from CrbnI391V/I391V mice degrade substrates when treated with thalidomide derivatives in vitro. We used mass spectrometry–based proteomics with TMT labeling of peptides for quantification30 to assess changes in the global proteome of c-Kit+ hematopoietic stem and progenitor cells from knock-in and wild-type mice treated in vitro with 1 µM lenalidomide for 12 hours. In CrbnI391V/I391V cells, lenalidomide treatment resulted in a selective and statistically significant decrease in the protein abundance of Ikzf1 (adjusted P= .02 for isoform CRA_a and adjusted P = .03 isoform CRA_b) and Ck1α (adjusted P = .02) relative to vehicle-treated cells (Figure 1A-B; supplemental Table 1). In contrast, no statistically significant changes in protein abundance were observed following lenalidomide treatment of wild-type cells.

Degradation of known thalidomide-derivative targets in CrbnI391Vmice. (A-C) c-Kit+ hematopoietic stem and progenitor cells were isolated from CrbnI391V/I391V or wild-type (WT) mice and treated in vitro with 1 µM lenalidomide, 1 µM pomalidomide, or DMSO vehicle for 12 hours. Protein abundance was assessed by liquid chromatography-mass spectrometry using TMT10 isobaric tagging reagents for quantification. Data are plotted as adjusted P value vs log2 fold change; the full range of log2 fold change values is shown as an inset. The horizontal line indicates an adjusted P value of .05 and proteins with a P value < .05 are shown in red. Log2 fold change is the average of 2 biological replicates. Ikzf3 is not expressed in c-Kit+ hematopoietic stem and progenitor cells and was not detected in these experiments. (D) Posttranslational degradation of an IKZF3 reporter in CrbnI391V/I391V or WT Hoxb8-transformed cells treated with lenalidomide, thalidomide, or pomalidomide in vitro for 6 hours. In this and other reporter constructs, an independently translated mCherry serves as a transcriptional control. (D-E) Posttranslational degradation of a ZFP91 flow reporter in CrbnI391V/I391V Hoxb8-transformed cells treated in vitro for 12 hours. (F) Degradation of Ikzf3 and CK1α as assessed by western blot in c-Kit+ and T cells from CrbnI391V/I391V,CrbnI391V/+, or WT mice after 18 hours of in vitro treatment with the drug indicated. All samples on the T-cell blot were run on adjacent lanes on the same gel, but the order of lanes has been changed for clarity. Results are representative of 3 independent experiments. (G) In vivo treatment of CrbnI391V/I391V mice with lenalidomide or thalidomide results in degradation of Ikzf1 and Ck1α in T cells after 12 hours. (H) T cells from CrbnI391V/I391V and CrbnI391V/+ mice have increased production of IL-2 by ELISA when treated with lenalidomide or pomalidomide for 24 hours, but no change in seen in WT cells. For WT cells, P value is not significant for all comparisons with DMSO. For CrbnI391V/+ and CrbnI391V/I391V, all comparisons with DMSO have a P value < .05. Statistical significance calculated with an unpaired Student t test and n ≥ 3. Results are representative of 2 independent experiments. Error bars are standard error of the mean. GFP, green fluorescent protein; IB, immunoblot; Len, lenalidomide; mIL, murine interleukin.
Degradation of known thalidomide-derivative targets in CrbnI391Vmice. (A-C) c-Kit+ hematopoietic stem and progenitor cells were isolated from CrbnI391V/I391V or wild-type (WT) mice and treated in vitro with 1 µM lenalidomide, 1 µM pomalidomide, or DMSO vehicle for 12 hours. Protein abundance was assessed by liquid chromatography-mass spectrometry using TMT10 isobaric tagging reagents for quantification. Data are plotted as adjusted P value vs log2 fold change; the full range of log2 fold change values is shown as an inset. The horizontal line indicates an adjusted P value of .05 and proteins with a P value < .05 are shown in red. Log2 fold change is the average of 2 biological replicates. Ikzf3 is not expressed in c-Kit+ hematopoietic stem and progenitor cells and was not detected in these experiments. (D) Posttranslational degradation of an IKZF3 reporter in CrbnI391V/I391V or WT Hoxb8-transformed cells treated with lenalidomide, thalidomide, or pomalidomide in vitro for 6 hours. In this and other reporter constructs, an independently translated mCherry serves as a transcriptional control. (D-E) Posttranslational degradation of a ZFP91 flow reporter in CrbnI391V/I391V Hoxb8-transformed cells treated in vitro for 12 hours. (F) Degradation of Ikzf3 and CK1α as assessed by western blot in c-Kit+ and T cells from CrbnI391V/I391V,CrbnI391V/+, or WT mice after 18 hours of in vitro treatment with the drug indicated. All samples on the T-cell blot were run on adjacent lanes on the same gel, but the order of lanes has been changed for clarity. Results are representative of 3 independent experiments. (G) In vivo treatment of CrbnI391V/I391V mice with lenalidomide or thalidomide results in degradation of Ikzf1 and Ck1α in T cells after 12 hours. (H) T cells from CrbnI391V/I391V and CrbnI391V/+ mice have increased production of IL-2 by ELISA when treated with lenalidomide or pomalidomide for 24 hours, but no change in seen in WT cells. For WT cells, P value is not significant for all comparisons with DMSO. For CrbnI391V/+ and CrbnI391V/I391V, all comparisons with DMSO have a P value < .05. Statistical significance calculated with an unpaired Student t test and n ≥ 3. Results are representative of 2 independent experiments. Error bars are standard error of the mean. GFP, green fluorescent protein; IB, immunoblot; Len, lenalidomide; mIL, murine interleukin.
Because lenalidomide and pomalidomide differ in their substrate specificities,17 we also evaluated the effect of 1 µM pomalidomide treatment on CrbnI391V/I391V c-Kit+ cells (Figure 1C; supplemental Table 2). We found that pomalidomide treatment decreased the protein abundance of Ikzf1 (adjusted P = .003 for isoform CRA_a and adjusted P = .003 for isoform CRA_b) relative to vehicle-treated cells. There was no statistically significant change in the protein level of Ck1α (adjusted P = .153), consistent with prior reports.17 Pomalidomide treatment also decreased the abundance of the zinc finger protein Zfp91 (adjusted P = .01), which was recently shown to be degraded by lenalidomide in human cells.31
To further examine the activity of thalidomide analogs in Crbn knock-in cells, we established an immortalized myeloid progenitor cell line by expressing the transcription factor Hoxb8 in c-Kit+ cells from CrbnI391V/I391V and wild-type mice.29 Using flow cytometry, we measured the posttranslational stability of an IKZF3 degron reporter in which a 60 amino acid thalidomide-sensitive region of human IKZF3 is fused in frame with GFP18 (supplemental Figure 2A). Treatment with low doses of lenalidomide, pomalidomide, and thalidomide induced degradation of the IKZF3 reporter in Hoxb8 cells from CrbnI391V/I391V mice (Figure 1D). Knock-in cells were ∼100-fold more sensitive to treatment with thalidomide derivatives than wild-type cells. No IKZF3 reporter degradation was seen in knock-in Hoxb8 cells expressing a mutant version of the IKZF3 degron that is resistant to thalidomide analogs (supplemental Figure 2B). Additionally, we found that thalidomide derivatives induced degradation of a similar reporter containing a pomalidomide-sensitive region of human ZFP91 in CrbnI391V/I391V but not wild-type Hoxb8 cells (Figure 1E).
We next asked whether treatment with thalidomide derivatives induces the degradation of endogenously expressed proteins. We found that ex vivo lenalidomide treatment of cultured splenic T cells and c-Kit+ bone marrow cells from CrbnI391V/I391V mice resulted in degradation of endogenous Ikzf3 and Ck1α, respectively (Figure 1F). No degradation of these proteins was observed with high-dose treatment of wild-type mouse cells, whereas CrbnI391V/+ cells had an intermediate phenotype.
We also observed degradation of endogenously expressed proteins following in vivo administration of thalidomide analogs (Figure 1G). Single-dose treatment with thalidomide derivatives decreased protein levels of Ikzf1, Ck1α, and Zfp91 in T cells from CrbnI391V/I391V mice (supplemental Figure 2C-D). Maximum degradation of these proteins was seen ∼6 hours after treatment with 50 mg/kg lenalidomide, with recovery of baseline protein levels by 24 hours. Although higher and more frequent doses of thalidomide derivatives are required to achieve a similar effect in mice as in humans, this is consistent with the higher rate of lenalidomide and thalidomide metabolism in mice.32-34
Thalidomide derivatives have immunomodulatory effects that are observed in humans but not mice, including increased production of the cytokine IL-2 by T cells.35 IKZF3 is a transcriptional repressor of the IL2 gene, and degradation of IKZF3 increases IL2 messenger RNA and protein.36 In CrbnI391V/+ and CrbnI391V/I391V T cells cultured in vitro and treated with lenalidomide or pomalidomide, we observed increased transcription and secretion of IL-2 (Figure 1H; supplemental Figure 2E). In contrast, no change in IL-2 transcription or secretion was observed in wild-type T cells treated with thalidomide derivatives.
In aggregate, these results demonstrate that endogenous expression of Crbn with a single I391V amino acid change is sufficient to confer thalidomide derivative–induced degradation of Ikzf1, Ikzf3, Ck1α, and Zfp91 in mouse cells, both in vivo and in vitro. Degradation of these substrates alters the biological activity of CrbnI391V cells, supporting the use of this model to study the activity of thalidomide derivatives in vivo.
Effect of lenalidomide treatment on native hematopoiesis
Much of the clinical efficacy and toxicity of thalidomide analogs is due to effects on hematopoietic cells that reside and differentiate within a complex microenvironment. We therefore examined the effects of lenalidomide, a drug that causes myelosuppression at high doses,37 on in vivo hematopoiesis. Administration of lenalidomide for 21 days resulted in a decline in total white blood cell count in CrbnI391V/I391V, but not wild-type mice, without a significant change in red blood cell (RBC) number (Figure 2A). Leukopenia was driven primarily by a decrease in the number of B220+ B cells, but there was also a statistically significant decrease in the number of CD3+ T cells and CD11b+ Gr1+ granulocytes. These findings are consistent with the role of IKZF1 and IKZF3 in lymphocyte development.38-40 We also observed a statistically significant decrease in the platelet counts of knock-in mice treated with lenalidomide, which was not seen in wild-type mice. This finding is consistent with thrombocytopenia seen in patients treated with lenalidomide, which is often a dose-limiting toxicity.5,11,12,41,42

Effect of lenalidomide treatment on hematopoiesis in CrbnI391Vmice. (A) Peripheral blood counts of WT and CrbnI391V/I391V mice after 21 days of treatment with 50 mg/kg lenalidomide or vehicle. CD11b+ Gr1+ granulocyte and platelet data are pooled from 2 treated cohorts of mice treated for 21 to 24 days. n ≥ 3 for data shown, which are representative of 4 independent experiments. (B) Hematopoietic stem and progenitor cell compartments in WT and CrbnI391V/I391V mice after 21 days of treatment with 50 mg/kg lenalidomide or vehicle. LK cells are Lineage− c-Kit+; ST-HSC are Lineage− c-Kit+ Sca1+ CD48− CD150−; LT-HSC are Lineage− c-Kit+ Sca1+ CD48− CD150+. n ≥ 3 for data shown, which are representative of 3 independent experiments. Lineage markers are CD3, B220, CD11b, Gr1, and Ter119. (C) Hematopoietic progenitor compartments in WT and CrbnI391V/I391V mice after 28 or 35 days of treatment with 50 mg/kg lenalidomide or vehicle. MPP are Lineage− c-Kit+ Sca1+ CD48+ CD150−; CMP are Lineage− c-Kit+ Sca1− CD34+ CD16/32mid; GMP are Lineage− c-Kit+ Sca1− CD34+ CD16/32hi; MEP are Lineage− c-Kit+ Sca1− CD34+ CD16/32low. Lineage markers are CD3, B220, CD11b, Ter119, and Gr1. n ≥ 5 for data shown, which are combined from 2 independent experiments. (D) Erythroid differentiation subsets as defined by CD71 and TER119 staining in the bone marrow from WT and CrbnI391V/I391V mice after 28 days of treatment with vehicle or 50 mg/kg lenalidomide. n ≥ 4 for data shown, which are representative of 3 independent experiments. **P < .005. P values calculated using unpaired Student t test. Error bars are standard error of the mean. LSK, Lineage− c-Kit+ Sca1+; ns, not significant; PLT, platelet; WBC, white blood cell.
Effect of lenalidomide treatment on hematopoiesis in CrbnI391Vmice. (A) Peripheral blood counts of WT and CrbnI391V/I391V mice after 21 days of treatment with 50 mg/kg lenalidomide or vehicle. CD11b+ Gr1+ granulocyte and platelet data are pooled from 2 treated cohorts of mice treated for 21 to 24 days. n ≥ 3 for data shown, which are representative of 4 independent experiments. (B) Hematopoietic stem and progenitor cell compartments in WT and CrbnI391V/I391V mice after 21 days of treatment with 50 mg/kg lenalidomide or vehicle. LK cells are Lineage− c-Kit+; ST-HSC are Lineage− c-Kit+ Sca1+ CD48− CD150−; LT-HSC are Lineage− c-Kit+ Sca1+ CD48− CD150+. n ≥ 3 for data shown, which are representative of 3 independent experiments. Lineage markers are CD3, B220, CD11b, Gr1, and Ter119. (C) Hematopoietic progenitor compartments in WT and CrbnI391V/I391V mice after 28 or 35 days of treatment with 50 mg/kg lenalidomide or vehicle. MPP are Lineage− c-Kit+ Sca1+ CD48+ CD150−; CMP are Lineage− c-Kit+ Sca1− CD34+ CD16/32mid; GMP are Lineage− c-Kit+ Sca1− CD34+ CD16/32hi; MEP are Lineage− c-Kit+ Sca1− CD34+ CD16/32low. Lineage markers are CD3, B220, CD11b, Ter119, and Gr1. n ≥ 5 for data shown, which are combined from 2 independent experiments. (D) Erythroid differentiation subsets as defined by CD71 and TER119 staining in the bone marrow from WT and CrbnI391V/I391V mice after 28 days of treatment with vehicle or 50 mg/kg lenalidomide. n ≥ 4 for data shown, which are representative of 3 independent experiments. **P < .005. P values calculated using unpaired Student t test. Error bars are standard error of the mean. LSK, Lineage− c-Kit+ Sca1+; ns, not significant; PLT, platelet; WBC, white blood cell.
We next examined the effects of lenalidomide on normal hematopoietic stem and progenitor cells, which is difficult to examine in patients with hematologic malignancies because of the presence of neoplastic hematopoiesis as well as therapy-induced alterations of the bone marrow microenvironment. Lenalidomide treatment resulted in a decrease in the proportion of Lineage− c-Kit+ and Lineage− c-Kit+ Sca1+ cells as well as phenotypically defined Lineage− c-Kit+ Sca1+ CD48− CD150+ long-term hematopoietic stem cells (LT-HSCs) and Lineage− c-Kit+ Sca1+ CD48− CD150− short-term hematopoietic stem cells (ST-HSCs) in the bone marrow (Figure 2B; supplemental Figure 3). We also observed a decrease in the proportion of multipotent progenitors (MPPs; Lineage− c-Kit+ Sca1+ CD48+ CD150−), common myeloid progenitors (CMPs; Lineage− c-Kit+ Sca1− CD34+ CD16/32intermediate), and granulocyte-macrophage progenitors (GMPs; Lineage− c-Kit+ Sca1− CD34+ CD16/32hi) in knock-in mice treated with lenalidomide, but not wild-type mice (Figure 2C; supplemental Figure 3). This indicates that long-term, high-dose treatment with lenalidomide impairs the hematopoietic stem and progenitor cell compartment, consistent with the myelosuppressive effects of high-dose lenalidomide.5,11,12,41,42
In contrast to its effects on other hematopoietic stem and progenitor cells, lenalidomide treatment did not significantly alter the percentage of megakaryocyte-erythroid progenitors (MEPs; Lineage− c-Kit+ Sca1− CD34+ CD16/32low; Figure 2C) in the bone marrow of CrbnI391V/I391V mice. Because the HSC, MPP, and CMP that give rise to the MEP lineage are all decreased in lenalidomide-treated CrbnI391V/I391V mice, this suggests that lenalidomide treatment enhances the development of the megakaryocyte-erythroid lineage, consistent with the normal RBC counts seen in these mice. We also observed expansion of the erythroblast II erythroid progenitor (Ter119+ CD71intermediate; Figure 2D) compartment in the bone marrow of lenalidomide-treated CrbnI391V/I391V mice, but not wild-type mice. Although the effects of lenalidomide or related analogs on normal erythropoiesis have not been examined previously in vivo, these effects are consistent with studies of lenalidomide on human erythroid progenitor cells cultured in vitro.43,44
In vivo modeling of efficacy of lenalidomide in cells haploinsufficient for Csnk1a1
We previously reported that lenalidomide induces the ubiquitination of CK1α, a protein expressed at haploinsufficient levels in del(5q) MDS, and proposed that the resulting degradation provides a mechanistic basis for lenalidomide’s therapeutic window in this condition.17 Here, we sought to use the CrbnI391V model to demonstrate that haploinsufficiency for Csnk1a1 is sufficient to explain lenalidomide’s therapeutic window in vivo.
To examine whether physiologic expression of CrbnI391V is sufficient to confer lenalidomide sensitivity to Csnk1a1+/− hematopoietic cells, we bred CrbnI391V mice to Csnk1a1 conditional knockout mice in which Cre recombinase is under the control of the Mx1 promoter.27 After inducing excision of the floxed Csnk1a1 allele in hematopoietic cells in vivo, we conducted a competitive in vitro culture experiment with congenic c-Kit+ hematopoietic stem and progenitor cells. We saw no effect of lenalidomide treatment on Crbn+/+Csnk1a1+/−MxCre+ cells (Figure 3A; supplemental Figure 4A-B). In contrast, CrbnI391VCsnk1a1+/−MxCre+ cells were significantly more sensitive to lenalidomide than CrbnI391VMxCre+ cells and were depleted from the culture over time (Figure 3A; supplemental Figure 4A-B). The depletion of CrbnI391VCsnk1a1+/−MxCre+ cells correlated with increased expression of the Tp53 transcriptional target p21 and an increased percentage of Annexin V+ cells, consistent with apoptotic cell death (supplemental Figure 4C-D). Lenalidomide-induced depletion was seen both in CrbnI391V/I391V and CrbnI391V/+ cells, confirming that heterozygous expression of CrbnI391V is sufficient to confer response.

CrbnI391Vcells heterozygous for Csnk1a1 are more sensitive to lenalidomide in vitro and in vivo. (A) An in vitro competition experiment was performed by isolating CD45.2+ c-Kit+ cells of the listed genotypes, mixing them in a 1:1 ratio with CD45.1+ c-Kit+ cells of the same Crbn genotype, treating the cells with vehicle or 10 μM lenalidomide, and following the percentage of CD45.1+ and CD45.2+ cells in the cultures over time by flow cytometry. The data are given as the ratio of the percentage of CD45.2+ cells to the percentage of CD45.1+ cells at each time point, normalized to the value of the DMSO-treated sample at that time point. n = 4. (B-C) Peripheral blood chimerism over time in transplanted mice treated with vehicle or 50 mg/kg lenalidomide twice per day. Data are given as a ratio of the percentage of CD45.2+ cells (test genotype) in the peripheral blood relative to the sum of the percentage of CD45.1+/CD45.2+ (competitor) and CD45.2+ (test genotype) cells. This method of data analysis excludes any CD45.1+ cells arising from residual recipient-derived hematopoiesis. (D) Chimerism in hematopoietic stem and progenitor compartments of transplanted mice after 41 days of treatment with vehicle or 50 mg/kg lenalidomide twice per day. Lineage markers are CD3, B220, CD11b, Gr1, and Ter119. Dark shading represents the vehicle and light shading represents lenalidomide. (E) Peripheral blood chimerism results for in vivo competition experiment with cells heterozygous for both Csnk1a1 and Rps14. Mice were treated twice per day with vehicle of 50 mg/kg lenalidomide for 41 days. Data are expressed as in panel B. For all in vivo experiments, n = 4 and results are representative of 2 independent experiments.
CrbnI391Vcells heterozygous for Csnk1a1 are more sensitive to lenalidomide in vitro and in vivo. (A) An in vitro competition experiment was performed by isolating CD45.2+ c-Kit+ cells of the listed genotypes, mixing them in a 1:1 ratio with CD45.1+ c-Kit+ cells of the same Crbn genotype, treating the cells with vehicle or 10 μM lenalidomide, and following the percentage of CD45.1+ and CD45.2+ cells in the cultures over time by flow cytometry. The data are given as the ratio of the percentage of CD45.2+ cells to the percentage of CD45.1+ cells at each time point, normalized to the value of the DMSO-treated sample at that time point. n = 4. (B-C) Peripheral blood chimerism over time in transplanted mice treated with vehicle or 50 mg/kg lenalidomide twice per day. Data are given as a ratio of the percentage of CD45.2+ cells (test genotype) in the peripheral blood relative to the sum of the percentage of CD45.1+/CD45.2+ (competitor) and CD45.2+ (test genotype) cells. This method of data analysis excludes any CD45.1+ cells arising from residual recipient-derived hematopoiesis. (D) Chimerism in hematopoietic stem and progenitor compartments of transplanted mice after 41 days of treatment with vehicle or 50 mg/kg lenalidomide twice per day. Lineage markers are CD3, B220, CD11b, Gr1, and Ter119. Dark shading represents the vehicle and light shading represents lenalidomide. (E) Peripheral blood chimerism results for in vivo competition experiment with cells heterozygous for both Csnk1a1 and Rps14. Mice were treated twice per day with vehicle of 50 mg/kg lenalidomide for 41 days. Data are expressed as in panel B. For all in vivo experiments, n = 4 and results are representative of 2 independent experiments.
To evaluate the selective effect of lenalidomide on Csnk1a1+/− hematopoietic cells in vivo, we performed a competitive transplantation assay with congenic markers (supplemental Figure 4E). We mixed CrbnI391V/I391VCsnk1a1+/+MxCre+ or CrbnI391V/I391VCsnk1a1fl/+MxCre+ cells with CrbnI391V/I391V competitor cells and transplanted a 1:1 mixture into lethally irradiated mice. After engraftment, we induced excision of the heterozygous floxed Csnk1a1 allele and treated the mice with lenalidomide or vehicle. In the peripheral blood, we observed a rapid and sustained depletion of Csnk1a1+/− cells in mice receiving lenalidomide, but not in those treated with vehicle (Figure 3B). Lenalidomide sensitivity of Csnk1a1+/− cells was apparent in all hematopoietic stem and progenitor subsets in the bone marrow, including phenotypically defined LT-HSC and ST-HSC (Figure 3D; supplemental Figure 4F-G), providing evidence of therapeutic activity of lenalidomide in hematopoietic stem cells.
Heterozygous deletion of Trp53 was sufficient to confer resistance to lenalidomide sensitivity in Csnk1a1+/− cells (Figure 3C). This resistance was seen in all hematopoietic stem and progenitor subsets, including LT-HSC, ST-HSC, GMP, CMP, and MEP (Figure 3D; supplemental Figure 4F-G) and is consistent with the outgrowth of TP53 mutant cells seen in del(5q) patients who develop lenalidomide resistance.45-47 These results demonstrate that Tp53 plays a key downstream role in lenalidomide sensitivity of Csnk1a1+/− cells in vivo.
Patients with del(5q) MDS typically have large deletions on 5q that result in the haploinsufficient expression of several genes. The ribosomal protein Rps14 is encoded within the del(5q) common deleted region, and heterozygous loss of Rps14 induces Tp53, resulting in an erythroid differentiation defect.28,48 We therefore investigated whether heterozygous loss of Rps14 increases lenalidomide sensitivity using Rps14 conditional knockout mice.28 To test whether Csnk1a1+/−Rps14+/− cells are more sensitive to lenalidomide than Csnk1a1+/− cells, we performed a competitive transplantation assay. As shown in Figure 3E, the percentage of CrbnI391V/I391VRps14+/−MxCre+ cells in the peripheral blood of transplanted mice was not altered by lenalidomide treatment. This suggests that, unlike Csnk1a1, deletion of a single allele of Rps14 does not confer increased lenalidomide sensitivity. CrbnI391V/I391VCsnk1a1+/−Rps14+/−MxCre+ cells were depleted from the peripheral blood with lenalidomide treatment, but this effect was indistinguishable from the lenalidomide-induced depletion of CrbnI391V/I391VCsnk1a1+/−MxCre+ cells. Thus, lenalidomide-induced depletion is driven by haploinsufficiency for Csnk1a1 without significant contribution from Rps14 haploinsufficiency.
Our results provide the first demonstration that haploinsufficiency for Ck1α is sufficient to explain the selective elimination of del(5q) cells in response to lenalidomide in vivo, and that the drug alters both the stem and progenitor compartments. We confirmed the essential role of Tp53 as a downstream effector in lenalidomide-induced selection against Csnk1a1+/− cells and as a driver of in vivo resistance at the stem cell level.
Thalidomide-induced fetal loss in CrbnI391V mice
The differences between species in CRBN sequence and response to thalidomide derivatives suggests a possible basis for the species-specific teratogenicity of thalidomide. Although there are >20 amino acid differences between mouse and human CRBN, the V388 residue in human CRBN, corresponding to residue to I391V in mice, correlates with thalidomide teratogenicity (Figure 4A; supplemental Table 3). In contrast to mice, rabbits carry a valine at this position and have thalidomide embryopathy.16,49,50 The only primate known to carry the mouse-like isoleucine residue, the greater bushbaby, is also the only primate known to be resistant to thalidomide.51 Other thalidomide-resistant species, including rats, cats, and pigs also have an isoleucine at this position.
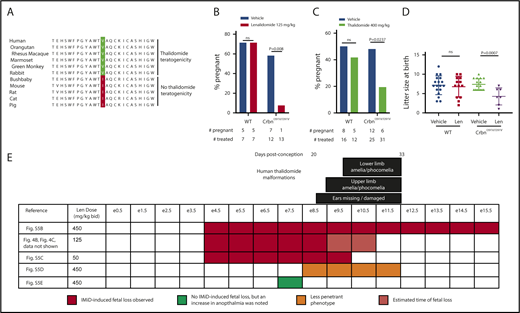
Thalidomide derivative–induced teratogenicity in CrbnI391Vmice. (A) Comparison of CRBN sequences in species with and without thalidomide teratogenicity. Thalidomide teratogenicity correlates with the presence of a valine at the amino acid position corresponding with 388 in human CRBN (highlighted), whereas the presence of an isoleucine at this position predicts lack of thalidomide teratogenicity. More details about the sequences included in the alignment are in supplemental Table 3. (B) Lenalidomide induces fetal loss of CrbnI391V/I391V litters but not wild-type litters. Plugged wild-type or CrbnI391V/I391V dams who had been mated with males of the same genotype were treated with 125 mg/kg lenalidomide every 12 hours from e4.5 to e10.5. Data shown are percent of dams pregnant at e17.5. The data at bottom lists the total number of dams treated and the number pregnant at e17.5. P values are from an N-1 χ2 test. (C) Thalidomide also induces fetal demise of CrbnI391V/I391V litters but not wild-type litters. Plugged wild-type or CrbnI391V/I391V dams who had been mated with males of the same genotype were treated with 400 mg/kg thalidomide from e4.5 to e10.5. Data shown are percent of dams pregnant at harvests on e10.5 and e17.5 combined. The data at bottom lists the total number of dams treated and the number pregnant. P values are from an N-1 χ2 test. (D) Effect of lenalidomide treatment on litter size at birth. Given that lenalidomide treatment of pregnant CrbnI391V/I391V dams causes a penetrant fetal loss phenotype, data are pool of multiple experiments in which dams were treated with 25 to 125 mg/kg of lenalidomide for e4.5 to e9.5 or e4.5 to e10.5 presumed days of gestation. (E) Determination of critical period for lenalidomide treatment in mice. Each row shows a treatment schedule, with the color reflecting whether this dose and treatment schedule fetal loss phenotype. Black bars at top represent an approximate comparison with the critical period for various thalidomide-induced malformations in humans, given in days postconception. Human critical periods are from Vargesson.1 Corresponding mouse embryonic stages were assigned by comparison with Carnegie stages.52,53 P values are from an unpaired Student t test. bid, twice per day; IMiD, immunomodulatory drug.
Thalidomide derivative–induced teratogenicity in CrbnI391Vmice. (A) Comparison of CRBN sequences in species with and without thalidomide teratogenicity. Thalidomide teratogenicity correlates with the presence of a valine at the amino acid position corresponding with 388 in human CRBN (highlighted), whereas the presence of an isoleucine at this position predicts lack of thalidomide teratogenicity. More details about the sequences included in the alignment are in supplemental Table 3. (B) Lenalidomide induces fetal loss of CrbnI391V/I391V litters but not wild-type litters. Plugged wild-type or CrbnI391V/I391V dams who had been mated with males of the same genotype were treated with 125 mg/kg lenalidomide every 12 hours from e4.5 to e10.5. Data shown are percent of dams pregnant at e17.5. The data at bottom lists the total number of dams treated and the number pregnant at e17.5. P values are from an N-1 χ2 test. (C) Thalidomide also induces fetal demise of CrbnI391V/I391V litters but not wild-type litters. Plugged wild-type or CrbnI391V/I391V dams who had been mated with males of the same genotype were treated with 400 mg/kg thalidomide from e4.5 to e10.5. Data shown are percent of dams pregnant at harvests on e10.5 and e17.5 combined. The data at bottom lists the total number of dams treated and the number pregnant. P values are from an N-1 χ2 test. (D) Effect of lenalidomide treatment on litter size at birth. Given that lenalidomide treatment of pregnant CrbnI391V/I391V dams causes a penetrant fetal loss phenotype, data are pool of multiple experiments in which dams were treated with 25 to 125 mg/kg of lenalidomide for e4.5 to e9.5 or e4.5 to e10.5 presumed days of gestation. (E) Determination of critical period for lenalidomide treatment in mice. Each row shows a treatment schedule, with the color reflecting whether this dose and treatment schedule fetal loss phenotype. Black bars at top represent an approximate comparison with the critical period for various thalidomide-induced malformations in humans, given in days postconception. Human critical periods are from Vargesson.1 Corresponding mouse embryonic stages were assigned by comparison with Carnegie stages.52,53 P values are from an unpaired Student t test. bid, twice per day; IMiD, immunomodulatory drug.
We treated mice with lenalidomide or thalidomide during pregnancy and found a high rate of fetal loss in CrbnI391V/I391V mice, without any effect on wild-type mice (Figure 4B,C). When fetuses survived to birth, CrbnI391V/I391V litters treated with lenalidomide were significantly smaller than those treated with vehicle (Figure 4D). We narrowed the critical period for this effect to e4.5 through e9.5 (Figure 4D; supplemental Figure 5A-E). Fetal loss was observed to occur by e10.5, and dead but nonresorbed fetuses were observed at this time point (Figure 4E; supplemental Figure 5A). We observed a trend toward an increased rate of anophthalmia, a malformation observed in thalidomide embryopathy in humans, in litters treated at e7.5 only (supplemental Figure 5F), but otherwise litters that were carried to term appeared to have normal morphology at birth and at weaning.
Carnegie developmental stages provide a method to compare the chronology of the development of different vertebrate embryos.52,53 By comparison of Carnegie stages, the critical period for fetal loss in lenalidomide-treated CrbnI391V/I391V mice precedes the critical period for phocomelia and amelia in humans (Figure 4E), suggesting that the CrbnI391V mice may better recapitulate thalidomide-induced spontaneous abortion occurring before the critical period for malformations. In aggregate, these results demonstrate a clear, statistically significant effect of lenalidomide and thalidomide on CrbnI391V mouse embryos.
Discussion
Here, we report the development and characterization of a mouse model with a single isoleucine to valine point mutation in Crbn. These mice respond to thalidomide derivatives, enabling the first in vivo studies of these clinically important drugs in mice. We confirmed the degradation of the known targets Ikzf1, Ikzf3, Ck1α, and Zfp91, as well as the downstream phenotypic consequences of degradation of these proteins in primary cells.
We provide the first in vivo demonstration that Csnk1a1+/− cells are more sensitive to lenalidomide treatment, indicating that haploinsufficiency for CK1α is sufficient to explain the therapeutic window of lenalidomide in del(5q) MDS. We showed that lenalidomide targets phenotypically defined stem cells with heterozygous expression of Ck1α, and that Tp53 loss causes lenalidomide resistance in these stem cell populations, providing a mechanistic explanation for the selection for TP53 mutant clones seen in lenalidomide-treated patients.45-47
In vivo study of the CrbnI391V mice offered insights into clinically relevant toxicities of thalidomide derivatives. Lenalidomide treatment of CrbnI391V mice decreased the number of white blood cells, LT-HSC, and ST-HSC, recapitulating the myelosuppressive effects of lenalidomide and demonstrating that lenalidomide influences hematopoietic stem cells. These effects are consistent with the role of Ikzf1 and Ck1α in the survival of HSC.27,40
We also demonstrate that the CrbnI391V mutation is sufficient to confer thalidomide-induced fetal loss. Although thalidomide is most infamous for its characteristic malformations, it also caused an increased rate of stillbirths and an ∼40% rate of infant mortality.1,54,55 There is evidence that thalidomide can cause spontaneous abortions in humans if received before the critical period for malformations or at high doses,1,56,57 and both spontaneous abortions and malformations are observed in multiple nonhuman primates.58-60 Phocomelia and amelia may be absent in our model because of additional species-specific differences in the sequence of downstream targets, the phenotypic consequences of their degradation, or their dependence on Crbn residues other than I391V.
Degradation of known thalidomide derivative–induced protein targets such as IKZF1, IZKF3, or CK1α cannot readily explain thalidomide embryopathy. Germ line inactivation of IKZF1 or IKZF3 is tolerated without increased fetal loss or malformations38,39 and homozygous knock-out of CK1α leads to fetal death before e6.5,61 which is significantly earlier than thalidomide derivative–induced fetal death in CrbnI391V mice (Figure 4E).
Thalidomide derivatives were developed clinically without an understanding of their biochemical mechanism or predictive preclinical models. However, the discovery of thalidomide derivatives’ mechanism of action has enabled an understanding of the molecular basis of their clinical activity and now the generation of a predictive mouse model. The CrbnI391V knock-in mouse provides a model in which to investigate the effect of thalidomide derivatives on complex in vivo systems that are not well modeled in vitro, such as stem cell function, the immune system, embryonic development, and behavior. Additionally, CrbnI391V mice provide an opportunity to evaluate the relative contributions of immune-mediated and cell-autonomous effects to drug efficacy, which has been an open question in the field.62 A thalidomide-sensitive mouse offers several practical and scientific advantages compared with primates or rabbit models, including well-validated experimental systems and routine genome engineering, which will facilitate mechanistic studies.
We anticipate that CrbnI391V mice will play an important role in the future development of thalidomide derivatives by promoting the identification of novel substrates and allowing hypothesis-driven preclinical testing of new indications and novel derivatives, as well as studies of biomarkers of response and biological mechanisms of resistance.
The original mass spectrometry data has been deposited to MassIVE (http://massive.ucsd.edu; identifier: MSV000082653).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank members of the B.L.E. laboratory for helpful discussions and feedback on the manuscript, Quinn Sievers for providing the reporter vectors, and D. Sykes and D. Scadden for providing the Hoxb8 retroviral vector and CHO cells.
This work was supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL082945), National Cancer Institute (5F30CA199988 [E.C.F.], P01CA108631, and P50CA206963), and National Institute of General Medical Sciences (T32GM007753) (E.C.F.); Howard Hughes Medical Institute; Edward P. Evans Foundation; and Henry and Marilyn Taub Foundation.
Authorship
Contribution: E.C.F. and B.L.E. initiated the project, designed the experiments, and wrote the paper, with input from the other authors; E.C.F., M.M., D.N.A., S.D.H., B.L., and M.C. performed mouse experiments; J.A.K., A.A.G., and A.T.N. created and performed experiments with the Hoxb8 cell lines; and N.D.U., T.S., D.R.M., and S.A.C. performed and analyzed the mass spectrometry–based proteomics experiments.
Conflict-of-interest disclosure: The model described is covered by a patent assigned to Brigham and Women’s Hospital and Broad Institute which is licensed to Celgene. B.L.E. receives research support from Celgene. The remaining authors declare no competing financial interests.
Correspondence: Benjamin L. Ebert, Dana-Farber Cancer Institute, 450 Brookline Ave, D1610A, Boston, MA 02215; e-mail: benjamin_ebert@dfci.harvard.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal