

Key Points
Platelets directly interact with and kill circulating Plasmodium parasites in patients with malaria to help control parasitemia.
In vitro platelet antiplasmodicidal activity against P knowlesi involves platelet–cell binding and intracellular accumulation of PF4.

Abstract
Platelets are understood to assist host innate immune responses against infection, although direct evidence of this function in any human disease, including malaria, is unknown. Here we characterized platelet–erythrocyte interactions by microscopy and flow cytometry in patients with malaria naturally infected with Plasmodium falciparum, Plasmodium vivax, Plasmodium malariae, or Plasmodium knowlesi. Blood samples from 376 participants were collected from malaria-endemic areas of Papua, Indonesia, and Sabah, Malaysia. Platelets were observed binding directly with and killing intraerythrocytic parasites of each of the Plasmodium species studied, particularly mature stages, and was greatest in P vivax patients. Platelets preferentially bound to the infected more than to the uninfected erythrocytes in the bloodstream. Analysis of intraerythrocytic parasites indicated the frequent occurrence of platelet-associated parasite killing, characterized by the intraerythrocytic accumulation of platelet factor-4 and terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick-end labeling of parasite nuclei (PF4+TUNEL+ parasites). These PF4+TUNEL+ parasites were not associated with measures of systemic platelet activation. Importantly, patient platelet counts, infected erythrocyte-platelet complexes, and platelet-associated parasite killing correlated inversely with patient parasite loads. These relationships, taken together with the frequency of platelet-associated parasite killing observed among the different patients and Plasmodium species, suggest that platelets may control the growth of between 5% and 60% of circulating parasites. Platelet–erythrocyte complexes made up a major proportion of the total platelet pool in patients with malaria and may therefore contribute considerably to malarial thrombocytopenia. Parasite killing was demonstrated to be platelet factor-4-mediated in P knowlesi culture. Collectively, our results indicate that platelets directly contribute to innate control of Plasmodium infection in human malaria.
Introduction
Platelets are the second most abundant cell of the circulation after red blood cells (RBCs) and the principle regulators of hemostasis. Platelets can also integrate host immune responses through production of immunomodulatory molecules and via cell-to-cell interactions with white blood cells (WBCs), and may have host-protective roles in infectious disease.1 Platelets are an abundant source of antimicrobial molecules,2,3 have broad-spectrum pathogen-killing activities,4-9 and are required for host-mediated pathogen control and host survival in some infectious disease models.10-13 Clinically, low platelet counts are often associated with a poor prognosis and increased risk for infection.14-16 However, to date, direct evidence that platelets contribute to host protection in any human infectious disease is lacking.
Malaria kills more than 400 000 people each year.17 Although most deaths are caused by Plasmodium falciparum, all Plasmodium species can cause severe and fatal infection.18-21 Malaria pathogenesis is driven primarily by parasite biomass22-24 and modulated by host innate and adaptive immune responses.25,26 Thrombocytopenia is common in all malarias and is a risk factor for mortality in African children with falciparum malaria,27 Southeast Asian adults and children with falciparum and vivax malaria,28 and adults with knowlesi malaria.21 However, the mechanisms leading to thrombocytopenia are not fully understood, and its subsequent effects on parasite biomass, disease control, or progression has yet to be quantified for any of the human Plasmodium species.
Indirect evidence from separate sources suggests that platelets may contribute to the host protection in malaria. This includes observations of human platelets directly binding to and killing P falciparum-infected RBCs (iRBCs) in culture, as well as reduced survival in Plasmodium-infected mice depleted of platelets.12,13,29-31 A mechanism for the direct killing of Plasmodium involves platelet factor-4 (PF4), an abundant antimicrobial protein secreted by platelets, that, on entering the cell via the Duffy antigen, a chemokine receptor expressed by RBCs,30 kills P falciparum parasites by selectively lyzing the parasite digestive vacuole.29 However, a recent study did not reproduce the parasite killing effect of platelets in P chabaudi-infected mice or P falciparum-iRBCs,32 showing the difficulties in using in vitro and in vivo disease models to study these phenomena33 and highlighting the need for additional research in people with malaria.34
No clinical studies have addressed the role of platelets in killing P falciparum parasites across the spectrum of clinical disease, nor their role in protection and pathogenesis in human malaria from nonfalciparum Plasmodium species. Here we characterized cell-to-cell platelet interactions in patients naturally infected with P falciparum, Plasmodium vivax, Plasmodium malariae, or Plasmodium knowlesi and examined relationships between platelet-associated parasite killing and parasite biomass. We also demonstrate the mechanism by which human platelets can kill P knowlesi, a second culturable human Plasmodium species.
Methods
Study participants
In Papua, patients with malaria attending the Mitra Masyarakat Hospital in Timika were enrolled between 2014 and 2016. This lowland forest region has perennial transmission of P falciparum, P vivax, and P malariae. In Sabah, patients were enrolled between 2012 and 2016 from 3 district hospitals in Kudat Division as part of concurrent prospective clinical studies.35 Sabah is an area of low malaria transmission, which during this period was co-endemic for P falciparum, P vivax, and the zoonotic parasite P knowlesi.
Criteria for enrolment in both cohorts included a blood film positive by microscopy for any Plasmodium species, including mixed infections in Papua, fever or history of fever in the last 48 hours, no major concurrent illness or comorbidity, and no prior antimalarial therapy in the preceding 24 hours. Patients were excluded if pregnant or lactating. In Papua, patients younger than 16 or older than 60 years or with a hemoglobin concentration of 7 g/dL or less were also excluded. Severe malaria was defined according to World Health Organization 2014 research criteria.20 Hospitalization was at the discretion of the treating clinician in Papua and was mandatory in all patients with malaria in Sabah. All patients with malaria were treated according to local guidelines, as described previously.20,36-38 Control patients were selected from visitors or relatives of patients with malaria, with no fever or history of fever in the preceding 14 days and with a blood film negative for malaria parasites.
Details of blood collection and analysis procedures are described in supplemental Materials, available on the Blood Web site.
Platelet binding to iRBCs and uRBCs
Blood samples were analyzed by flow cytometry to measure platelet-bound iRBCs or uninfected RBCs (uRBCs), and expressed as frequencies and absolute numbers. In Papua, assays were performed on fresh samples, and in Sabah, on fixed blood (Cytofix, BD Biosciences). Fluorescent antibody panels comprised anti-CD45 (white cell exclusion marker), anti-CD41 or CD42b (platelet marker), anti-CD235ab (RBC marker), and either Hoechst 33342 or DRAQ-5 (parasite nuclear marker). The gating strategy for measuring platelet–iRBC/platelet–uRBC complexes in Papua is illustrated in Figure 1B and in Sabah in supplemental Figure 1A. Refer to supplemental Materials for details of the staining materials and procedures.
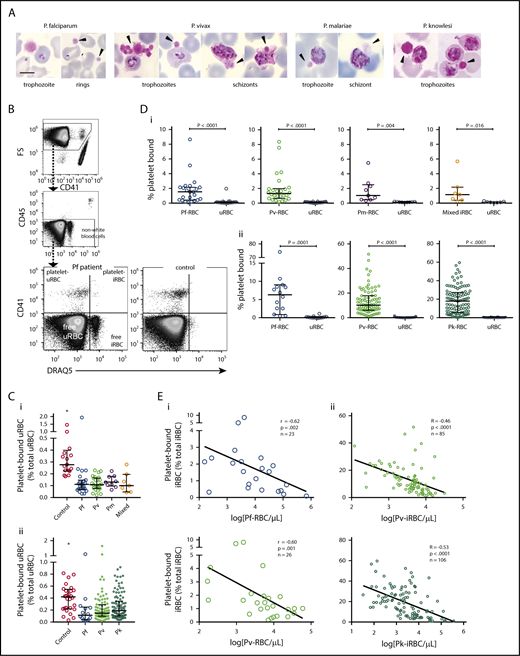
Platelet binding in clinical malaria blood samples. (A) Photos of platelet-bound iRBCs from patient Giemsa smears (black arrowhead = platelet). Images were taken at 1000× magnification using a Samsung Note-3 camera attached to an Olympus CX31 microscope. Scale bar, 5 µm. (B) Representative flow cytometry gating strategy to measure platelet binding in a P falciparum patient and healthy control. (C) Frequency of platelet-bound uRBCs by flow cytometry in patients with malaria compared with control patients in samples from (Ci) Papua (control patients, n = 17; Pf, n = 23; Pv, n = 26; Pm, n = 9; mixed, n = 7) and (Cii) Sabah (control patients, n = 28; Pf, n = 14; Pv, n = 85; Pk, n = 106; Kruskal-Wallis, *significantly different to all other groups). (D) Frequency of platelet-bound iRBCs and uRBCs by flow cytometry in samples from (Di) Papua (n as per Ci) and (Dii) Sabah (n as per Cii; Wilcoxon test). (E) Inverse correlation of platelet-bound iRBCs with parasitemia in samples from (Ei) Papua and (Eii) Sabah (Spearman). Scatterplots indicate median ± interquartile range for each group. Parasitemia values are log transformed. Data presented in supplemental Table 1. Pf, P falciparum; Pv, P vivax; Pm, P malariae; Pk, P knowlesi.
Platelet binding in clinical malaria blood samples. (A) Photos of platelet-bound iRBCs from patient Giemsa smears (black arrowhead = platelet). Images were taken at 1000× magnification using a Samsung Note-3 camera attached to an Olympus CX31 microscope. Scale bar, 5 µm. (B) Representative flow cytometry gating strategy to measure platelet binding in a P falciparum patient and healthy control. (C) Frequency of platelet-bound uRBCs by flow cytometry in patients with malaria compared with control patients in samples from (Ci) Papua (control patients, n = 17; Pf, n = 23; Pv, n = 26; Pm, n = 9; mixed, n = 7) and (Cii) Sabah (control patients, n = 28; Pf, n = 14; Pv, n = 85; Pk, n = 106; Kruskal-Wallis, *significantly different to all other groups). (D) Frequency of platelet-bound iRBCs and uRBCs by flow cytometry in samples from (Di) Papua (n as per Ci) and (Dii) Sabah (n as per Cii; Wilcoxon test). (E) Inverse correlation of platelet-bound iRBCs with parasitemia in samples from (Ei) Papua and (Eii) Sabah (Spearman). Scatterplots indicate median ± interquartile range for each group. Parasitemia values are log transformed. Data presented in supplemental Table 1. Pf, P falciparum; Pv, P vivax; Pm, P malariae; Pk, P knowlesi.
Platelet-associated parasite killing
Immunofluorescent microscopy of Cytofix-treated blood was used to quantify platelet-associated parasite killing, based on a terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick-end labeling (TUNEL) assay described elsewhere30 that labels degraded or sheared DNA, an indication of apoptosis or necrosis. Platelets and intraerythrocytic localization of PF4 were detected with an anti-human PF4 antibody (Abcam, United Kingdom), and intraerythrocytic Plasmodium DNA was identified using 4′,6-diamidino-2-phenylindole. Only high-parasitemia samples (>5000 parasites/µL) were analyzed and the number of parasites counted per sample recorded. In a subset of patients, iRBCs were distinguished into rings (4′,6-diamidino-2-phenylindole-positive without parasite pigment) or mature stages (4′,6-diamidino-2-phenylindole-positive with parasite pigment) by immunofluorescence, and the proportion of stages confirmed with Giemsa-stained blood smears performed in parallel. Refer to supplemental Materials for more detail.
Platelet activation
In Papua, platelet activation markers including platelet PAC-1 binding (recognizing the glycoprotein IIb/IIIa complex of activated platelets), P-selectin (CD62P) surface expression, and platelet–monocyte and platelet–neutrophil aggregate formation were measured using 2 flow cytometry panels. Plasma PF4 concentrations were measured by enzyme-linked immunosorbent assay. In Sabah, platelet–WBC aggregates were determined from the RBC-binding panel. The platelet activation gating strategies are illustrated in supplemental Figure 1B-C. Refer to supplemental Materials for details of the staining and analysis procedures.
In vitro parasite and platelet experiments
P knowlesi and P falciparum were cultured in 2.5% hematocrit O+ human RBCs, as described elsewhere.39,40 Platelets were purified from freshly collected human blood, as described previously.12 Refer to supplemental Materials for details of the culture conditions and platelet preparation.
Platelet count and quiescence were determined using an ADVIA 2120i hematology analyzer (Siemens, Australia). Platelet activation capacity was tested by treating platelets with 1 U/mL human thrombin for 10 minutes and then measuring PAC-1 binding and P-selectin surface expression by flow cytometry. All platelet preparations in this study (8 preparations from 6 donors) satisfied the criteria for quiescence (mean platelet volume, <9 fL and mean platelet component, >23 g/dL) and activation capacity (>1000-fold and >250-fold increase in PAC-1 and CD62P MFI, respectively, compared with untreated rested platelets; supplemental Figure 4).
For the parasite–platelet coculture experiments, P falciparum parasites were synchronized for ring stages the day before experiments, using 5% (wt/vol) d-sorbitol.41 Late-stage P knowlesi and P falciparum trophozoites were purified on the day of experiment, using 70% Percoll gradient centrifugation (1950g without brake for 10 minutes), and washed twice with complete culture medium. Parasitemia was adjusted to 0.5% at 2% hematocrit and platelets added; equal volumes of Tyrode’s buffer were added to untreated control wells. In some experiments, before adding to the parasites, recombinant human PF4 (PeproTech) and platelet lysates were incubated with 0.5 mg/mL preservative-free rabbit anti-human PF4 Ab (Abcam) or preservative-free rabbit serum (Novus Biologicals) for 15 minutes at 4°C. For transwell experiments, parasites were separated from 50 million platelets/mL, platelet lysate, or 0.5 μM PF4, using transwell inserts with 0.2 μm pore size (Anopore membrane, Nalge Nunc International, Denmark). The well inserts were presoaked with complete culture medium before use. P knowlesi was harvested after 24-hour and P falciparum after 48-hour culture with platelets, lysate, or PF4. Giemsa-stained thin smears were prepared, and 1000 RBCs were counted per slide at 1000× magnification. Percentage parasite growth was calculated using this formula: % parasite growth = [(treatment parasitemia − initial parasitemia) ÷ (untreated parasitemia − initial parasitemia)] × 100.
Platelet binding to parasites was determined in 24-hour cultures containing 2% asynchronous parasites and 60 million platelets/mL. Harvested cocultures were fixed in diluted Cytofix (Biosciences, Australia) for at least 24 hours and stored at 4°C. For flow cytometry quantitation, fixed cells were washed once with 1% (wt/vol) bovine serum albumin/phosphate-buffered saline and then stained with mouse anti-human CD42b conjugated to phycoerythrin and mouse anti-human CD235ab conjugated to allophycocyanin for 20 minutes at 4°C, followed by 5 µg/mL Hoechst 33342 for 5 minutes at 4°C. Fluorescence signals were measured using the LSR Fortessa (BD Biosciences). At least 100 000 events were collected per sample.
Statistics
The Mann-Whitney test or the Kruskal-Wallis test followed by Dunn’s multiple comparisons was used for between-group comparisons of clinical data. The Wilcoxon matched-pairs signed rank test was applied to paired datasets. One-way ANOVA with Sidak’s multiple comparisons was used for in vitro data. Associations between 2 variables were assessed using Spearman correlation. Bonferroni corrections to P values were applied where appropriate. Data were log transformed for multivariate comparisons. All statistical analyses were performed using GraphPad Prism 7 (GraphPad Software, La Jolla, CA) or Stata 14 (StataCorp, TX).
Study approval
The study was approved by the Human Research Ethics Committees of Gadjah Mada University, Indonesia (Papua), the Malaysian Ministry of Health (Sabah) (NMRR 10-754-6684), Menzies School of Health Research, Australia (Papua and Sabah), the Macquarie University, and the Australian National University. Written informed consent was obtained from all participants.
Results
Study participants
Separate cohorts of patients with malaria and healthy control patients were enrolled in Papua, Indonesia (n = 143), and Sabah, Malaysia (n = 233). Patients were categorized according to Plasmodium species and baseline characteristics, including parasitemia, platelet counts, and disease severity presented (Tables 1 and 2). The majority of patients with malaria at each site had nonsevere malaria: 92% in Papua and 97% in Sabah. Relative to control patients, platelet counts were significantly lower in malaria from all Plasmodium species at both study sites and correlated inversely with parasitemia in the P vivax groups (Papua, r = −0.37; P = .022; n = 38; and Sabah, r = −0.36; P = .0008; n = 84) and with histidine-rich protein-2 in falciparum malaria in Papua (r = −0.45; P = .0007; n = 54).
Baseline characteristics of 143 patients with malaria and healthy control patients from Papua
| Control patients (n = 24) | P falciparum (n = 55) | P vivax (n = 38) | P malariae (n = 14) | Mixed infection (n = 12)* | Kruskal-Wallis test P value | |
|---|---|---|---|---|---|---|
| Age, y | 28 (26-33.8) | 29 (19-37) | 24 (19-32.8) | 33.5 (26-47) | 22 (19.3-33) | .027 |
| Males, n (%) | 10 (42%) | 29 (53%) | 23 (61%) | 4 (29%) | 7 (58%) | — |
| Ethnicity, Highland/Lowland/non-Papuan, n | 12/11/1 | 50/2/3 | 33/3/2 | 14/0/0 | 12/0/0 | — |
| Number with severe malaria† | — | 8 | 1 | — | — | — |
| Parasitemia, parasites/µL | Not detected by microscopy | 15 600 (3660-105 000)‡ | 6160 (2120-11 400) | 1230 (815-2540) | 7160 (4590-106 000) | <.0001 |
| Hemoglobin, g/dL | 12.3 (11.5-14.6) | 12.4 (10.2-13.4) | 11.9 (10.5-13.1) | 9.6** (8.6-10.6) | 11.7 (8.8-13.3) | .0007 |
| WBC count, ×1000/µL | 7.4 (6.2-8.2) | 5.1** (3.7-6.6) | 5.5 (4.8-7.5) | 4.2** (3.2-5.1) | 5.8 (4.4-7.2) | <.0001 |
| Monocyte count, ×1000/µL | 0.45 (0.42-0.55) | 0.53 (0.30-0.77) | 0.54 (0.40-0.67) | 0.54 (0.37-0.68) | 0.51 (0.20-0.66) | .780 |
| Neutrophil count, ×1000/µL | 3.5 (2.5-4.4) | 2.8 (2.0-4.8) | 3.8 (2.8-4.3) | 1.6* (1.5-2.4) | 3.9 (2.6-4.8) | .0006 |
| Platelet count, ×1000/µL | 202 (171-228) | 66** (39-99)¶ | 78** (53-113) | 80** (56-101) | 60** (44-120) | <.0001 |
| Control patients (n = 24) | P falciparum (n = 55) | P vivax (n = 38) | P malariae (n = 14) | Mixed infection (n = 12)* | Kruskal-Wallis test P value | |
|---|---|---|---|---|---|---|
| Age, y | 28 (26-33.8) | 29 (19-37) | 24 (19-32.8) | 33.5 (26-47) | 22 (19.3-33) | .027 |
| Males, n (%) | 10 (42%) | 29 (53%) | 23 (61%) | 4 (29%) | 7 (58%) | — |
| Ethnicity, Highland/Lowland/non-Papuan, n | 12/11/1 | 50/2/3 | 33/3/2 | 14/0/0 | 12/0/0 | — |
| Number with severe malaria† | — | 8 | 1 | — | — | — |
| Parasitemia, parasites/µL | Not detected by microscopy | 15 600 (3660-105 000)‡ | 6160 (2120-11 400) | 1230 (815-2540) | 7160 (4590-106 000) | <.0001 |
| Hemoglobin, g/dL | 12.3 (11.5-14.6) | 12.4 (10.2-13.4) | 11.9 (10.5-13.1) | 9.6** (8.6-10.6) | 11.7 (8.8-13.3) | .0007 |
| WBC count, ×1000/µL | 7.4 (6.2-8.2) | 5.1** (3.7-6.6) | 5.5 (4.8-7.5) | 4.2** (3.2-5.1) | 5.8 (4.4-7.2) | <.0001 |
| Monocyte count, ×1000/µL | 0.45 (0.42-0.55) | 0.53 (0.30-0.77) | 0.54 (0.40-0.67) | 0.54 (0.37-0.68) | 0.51 (0.20-0.66) | .780 |
| Neutrophil count, ×1000/µL | 3.5 (2.5-4.4) | 2.8 (2.0-4.8) | 3.8 (2.8-4.3) | 1.6* (1.5-2.4) | 3.9 (2.6-4.8) | .0006 |
| Platelet count, ×1000/µL | 202 (171-228) | 66** (39-99)¶ | 78** (53-113) | 80** (56-101) | 60** (44-120) | <.0001 |
All values are median (interquartile range) unless otherwise indicated. Kruskal-Wallis with Dunn’s multiple comparisons test, significantly different from control patients (**P < .0005; *P < .005).
Mixed infection of P falciparum and P vivax.
Severe malaria criteria encountered: cerebral malaria (Glasgow Coma Score ≤10 for >30 minutes), jaundice (visible jaundice OR creatinine >1.5 mg/dL OR bilirubin >3 mg/dL AND >100 000 parasites/µL), acute renal failure (creatinine >3 mg/dL ± urine output <400 mL/day OR urea >20 mM), hypoglycemia (plasma glucose <40 mg/dL), hyperparasitemia (asexual parasitemia >10%), hypotension (blood pressure <80 mm Hg AND cool peripheries), and respiratory distress (respiratory rate >30/minute AND O2 saturation <92%).
Median parasitemia in severe (283 000 parasites/µL) vs nonsevere P falciparum (10 800 parasites/µL; Mann-Whitney test P = .0009).
Median platelet count in severe (18 500/µL) vs nonsevere P falciparum (74 000/µL; Mann-Whitney test P < .0001).
Baseline characteristics of 233 patients with malaria and healthy control patients from Sabah
| Control patients (n = 28) | P falciparum (n = 14) | P vivax (n = 85) | P knowlesi (n = 106) | Kruskal-Wallis test P value | |
|---|---|---|---|---|---|
| Age, y | 31.5 (26-36.8) | 25 (18-37.8) | 22 (12-34)** | 34 (22-52.3) | <.0001 |
| Number aged ≤12 y | — | — | 22 | 6 | — |
| Males, n (%) | 10 (36%) | 12 (86%) | 62 (73%) | 84 (79%) | — |
| Number with severe malaria* | — | 1 | 0 | 6 | — |
| Parasitemia, parasites/µL | Not detected by PCR | 9850 (3130-21 800) | 4640 (1830-7630) | 2650 (648-8770)† | .025 |
| Hemoglobin, g/dL | 13.7 (12.9-14.8) | 12.9 (11.1-14.8) | 12.4** (10.5-13.9) | 13.4 (11.9-14.6) | .002 |
| RBC count, ×106/ µL | 5.1 (4.9-5.5) | 4.9 (4.2-5.3) | 4.6 (4.1-5.0)*** | 5.1 (4.6-5.5) | <.0001 |
| WBC count, ×1000/µL | 7.8 (6.6-8.9) | 5.7** (4.1-6.9) | 6.4* (5.3-7.9) | 6.2** (5.1-7.9) | .002 |
| Platelet count, ×1000/µL | 348 (267-363) | 86*** (56-156) | 97*** (67-133) | 72*** (52-108)‡ | <.0001 |
| Control patients (n = 28) | P falciparum (n = 14) | P vivax (n = 85) | P knowlesi (n = 106) | Kruskal-Wallis test P value | |
|---|---|---|---|---|---|
| Age, y | 31.5 (26-36.8) | 25 (18-37.8) | 22 (12-34)** | 34 (22-52.3) | <.0001 |
| Number aged ≤12 y | — | — | 22 | 6 | — |
| Males, n (%) | 10 (36%) | 12 (86%) | 62 (73%) | 84 (79%) | — |
| Number with severe malaria* | — | 1 | 0 | 6 | — |
| Parasitemia, parasites/µL | Not detected by PCR | 9850 (3130-21 800) | 4640 (1830-7630) | 2650 (648-8770)† | .025 |
| Hemoglobin, g/dL | 13.7 (12.9-14.8) | 12.9 (11.1-14.8) | 12.4** (10.5-13.9) | 13.4 (11.9-14.6) | .002 |
| RBC count, ×106/ µL | 5.1 (4.9-5.5) | 4.9 (4.2-5.3) | 4.6 (4.1-5.0)*** | 5.1 (4.6-5.5) | <.0001 |
| WBC count, ×1000/µL | 7.8 (6.6-8.9) | 5.7** (4.1-6.9) | 6.4* (5.3-7.9) | 6.2** (5.1-7.9) | .002 |
| Platelet count, ×1000/µL | 348 (267-363) | 86*** (56-156) | 97*** (67-133) | 72*** (52-108)‡ | <.0001 |
All values are median (interquartile range) unless otherwise indicated. Kruskal-Wallis with Dunn’s multiple comparisons test, significantly different to control patients (***P < .0005; **P < .005; *P < .05).
Severe malaria criteria encountered: acute kidney injury (creatinine > 265 µmol/L), significant abnormal bleeding, severe anemia (hemoglobin <7 g/dL or hematocrit <20%), jaundice (visible jaundice OR creatinine >1.5 mg/dL OR bilirubin >3 mg/dL AND >100 000 parasites/µL [P falciparum] or >20 000/µL [P knowlesi]), hyperparasitemia (asexual parasitemia >10% [P falciparum] OR >100 000/µL [P knowlesi]), metabolic acidosis (HCO3 <15 mmol/L OR lactate >5 mmol/L).
Median parasitemia in severe (136 000 parasites/µL) vs nonsevere P knowlesi (2450 parasites/µL; Mann-Whitney test, P < .0001).
Median platelet count in severe (52 500/µL) vs nonsevere P knowlesi (72 000/µL; Mann-Whitney test, P = .154).
Platelet–iRBC and platelet–uRBC complexes are formed in the circulation of patients with malaria
Examination of patient thin blood smears revealed platelet binding to both iRBCs and uRBCs. Platelets were bound to all asexual parasite stages in each of the Plasmodium species, with evidence of platelet aggregates surrounding P vivax-iRBCs (Figure 1A). Characteristic features of dying parasites within platelet-bound iRBCs were observed, including spread of parasite pigment, suggesting dissolution of the digestive vacuole.
Flow cytometry was used to quantify the proportions of uRBCs and iRBCs that were each bound to platelets, hereafter platelet–uRBC and platelet–iRBC complexes (Figure 1B; supplemental Figure 1A). Proportions of platelet–uRBC complexes were similar among all the patient groups (median range, 0.10%-0.19%) and were significantly lower than the control patients (0.28% and 0.42%; Figure 1Ci and ii, respectively). In contrast, greater proportions of platelet–iRBCs were observed in all the patient groups (Figure 1Di-ii; supplemental Table 1). The greatest proportions were observed in P knowlesi patients (median, 18.1%), which were significantly higher than those in Sabah P falciparum patients (median, 6.3%; P = .002; supplemental Table 1). Overall complex magnitudes were lower in the Papua than the Sabah groups, which may be attributable to differences in sample preparation and staining. However, for all groups, platelet–iRBC complexes were significantly greater than platelet–uRBC complexes. Compared as a ratio, the proportions of platelet–iRBC complexes were between 8- and almost 100-fold greater than platelet–uRBC complexes (supplemental Table 1). We also separately analyzed the subgroup of 28 Sabah patients with P vivax or P knowlesi malaria who were children aged 12 years or younger (Table 2); similar proportions of platelet–iRBC/platelet–uRBC complexes to those in adults, and significantly greater frequencies of platelet–iRBC vs platelet–uRBC complexes, were observed in these groups (supplemental Figure 3A-C).
Using the proportions of the complexes and the respective RBC count in each individual, we calculated concentrations of platelet–RBC complexes (iRBCs and uRBCs combined). The medians ranged between 3100 and 13 600/µL blood (Table 3). Compared with respective circulating levels of free platelets (ie, free platelet:platelet–RBC complex ratio; Table 3), the complexes comprise a substantial proportion of the total platelet pool. For example, in P knowlesi-infected patients, on average, 1 complex was predicted to exist for every 7 noncomplexed platelets. The highest ratio, in P malariae, predicted 1 complex for every 19 platelets (Table 3). By comparison, free platelet:platelet–WBC ratios were much higher (≥118), indicating these complexes were only a minor component of the platelet pool (Table 3).
Circulating platelet–RBC and platelet–WBC concentrations and ratios to free platelets
| Patient cohort and Plasmodium species | Samples analyzed, n | Platelet–RBC complexes | Platelet–WBC complexes | ||
|---|---|---|---|---|---|
| ×103/µL blood* | Free platelet: complex ratio† | ×103/µL blood‡ | Free platelet: complex ratio¶ | ||
| Papua | |||||
| Control patients | 17 | 13.6 (10.1-20.9) | 14.5 (10.3-18.8) | 0.3 (0.2-0.4) | 713 (425-1146) |
| P falciparum | 23 | 4.5 (3.0-7.2) | 16.6 (13.2-22.5) | 0.2 (0.1-0.4) | 237 (147-451)*** |
| P vivax | 26 | 4.8 (3.0-7.7) | 17.2 (12.7-21.1) | 0.2 (0.2-0.3) | 393 (203-679)* |
| P malariae | 9 | 4.9 (3.6-6.5) | 18.5 (16.2-22.1) | 0.2 (0.1-0.3) | 348 (199-674) |
| Mixed | 7 | 3.1 (2.5-9.7) | 17.9 (14.1-18.7) | 0.3 (0.1-0.5) | 287 (138-345)* |
| P value§ | <.0001 | .561 | .663 | .009 | |
| Sabah | |||||
| Control patients | 27 | 21.4 (11.2-27.6) | 18.2 (11.8-35.2) | 1.1 (0.8-1.4) | 316 (216-448) |
| P falciparum | 12 | 6.0 (2.8-11.3)** | 14.6 (7.5-34.5) | 0.4 (0.1-0.6)**** | 391 (203-998) |
| P vivax | 78 | 6.8 (4.4-13.9)**** | 14.0 (6.1-26.2) | 0.8 (0.5-1.4) | 124 (66-209)**** |
| P knowlesi | 92 | 10.5 (5.4-18.3)* | 7.3 (4.0-13.2)**** | 0.6 (0.3-1.0)** | 118 (70-283)**** |
| P value§ | <.0001 | <.0001 | <.0001 | .004 | |
| Patient cohort and Plasmodium species | Samples analyzed, n | Platelet–RBC complexes | Platelet–WBC complexes | ||
|---|---|---|---|---|---|
| ×103/µL blood* | Free platelet: complex ratio† | ×103/µL blood‡ | Free platelet: complex ratio¶ | ||
| Papua | |||||
| Control patients | 17 | 13.6 (10.1-20.9) | 14.5 (10.3-18.8) | 0.3 (0.2-0.4) | 713 (425-1146) |
| P falciparum | 23 | 4.5 (3.0-7.2) | 16.6 (13.2-22.5) | 0.2 (0.1-0.4) | 237 (147-451)*** |
| P vivax | 26 | 4.8 (3.0-7.7) | 17.2 (12.7-21.1) | 0.2 (0.2-0.3) | 393 (203-679)* |
| P malariae | 9 | 4.9 (3.6-6.5) | 18.5 (16.2-22.1) | 0.2 (0.1-0.3) | 348 (199-674) |
| Mixed | 7 | 3.1 (2.5-9.7) | 17.9 (14.1-18.7) | 0.3 (0.1-0.5) | 287 (138-345)* |
| P value§ | <.0001 | .561 | .663 | .009 | |
| Sabah | |||||
| Control patients | 27 | 21.4 (11.2-27.6) | 18.2 (11.8-35.2) | 1.1 (0.8-1.4) | 316 (216-448) |
| P falciparum | 12 | 6.0 (2.8-11.3)** | 14.6 (7.5-34.5) | 0.4 (0.1-0.6)**** | 391 (203-998) |
| P vivax | 78 | 6.8 (4.4-13.9)**** | 14.0 (6.1-26.2) | 0.8 (0.5-1.4) | 124 (66-209)**** |
| P knowlesi | 92 | 10.5 (5.4-18.3)* | 7.3 (4.0-13.2)**** | 0.6 (0.3-1.0)** | 118 (70-283)**** |
| P value§ | <.0001 | <.0001 | <.0001 | .004 | |
All values are median (interquartile range) unless otherwise indicated.
Formula = ([%platelet–uRBCs from flow data] ÷ 102 × [RBC count from analyzer] × 106) + ([%platelet–iRBCs from flow data] ÷ 102 × [parasites/ µL]).
Formula = (platelet count from analyzer × 103) ÷ (platelet–RBCs/µL*).
Formula = ([%platelet-WBCs from flow data] ÷ 102) × (WBC count from analyzer) × 103.
Formula = (platelet count from analyzer × 103) ÷ (platelet–WBCs/µL‡).
Kruskal-Wallis with Dunn’s multiple comparisons test, significantly different from control patients (****P < .0001; ***P < .0005; **P < .005; *P < .05).
Correlative analyses revealed inverse relationships between platelet–iRBC complexes and parasitemia; these were significant in malaria from each species except P malariae (Figure 1Ei-ii; supplemental Figure 2A). After adjusting for platelet count, these relationships remained significant for P vivax and P knowlesi (both P < .0001). No significant relationships were observed between parasitemia and platelet–uRBC complexes. There was no consistent relationship between complexes and hemoglobin levels.
Taken together, these data show that platelets bind and form stable complexes with RBCs, with a greater preference for iRBCs than uRBCs. The frequencies of these complexes in the circulation of patients are substantial and compared with free platelets comprise a relatively large proportion of the total platelet pool. In addition, the proportions of platelet–iRBC complexes are higher in patients with low levels of parasites, and lower in patients with high parasite burden.
Platelets directly kill parasites in the circulation of patients with malaria
We conducted TUNEL and PF4 immunostaining on patient blood to detect and quantify the frequencies of dead intraerythrocytic parasites and the co-occurrence of PF4 accumulation within the iRBCs. With these methods, iRBCs containing PF4 and TUNEL labeling (PF4+TUNEL+) are indicative of direct platelet contact with the cell (by uptake of platelet-produced PF4) and consequent PF4-mediated killing of the parasite.29,30 We observed PF4+TUNEL+ iRBCs in all Plasmodium species in both patient cohorts (Figure 2A). Quantification of these observations showed substantial variances in the proportions of PF4+TUNEL+ iRBCs, although the medians calculated for each species were similar, except for P vivax, which was significantly greater in both Papua and Sabah compared with the other species (Figure 2B; Table 4). Notably, PF4 was present in more than half of the TUNEL+ (dead) parasites; in Papua, a median of 96% of TUNEL+P vivax-iRBCs contained PF4, which was significantly greater than the 52% median observed in P falciparum (P < .0001) and 69% median in P malariae (P = .051; Figure 2C). No PF4 staining or TUNEL labeling was observed in enriched suspensions of uninfected reticulocytes. The proportion of PF4+TUNEL+ iRBCs correlated inversely with parasitemia in Papuan patients infected with P vivax (r = −0.42; P = .020) and P falciparum (r = −0.36; P = .010); no significant relationships were observed in the other cohort groups, in which fewer samples were examined (Figure 2D; supplemental Figure 2B).
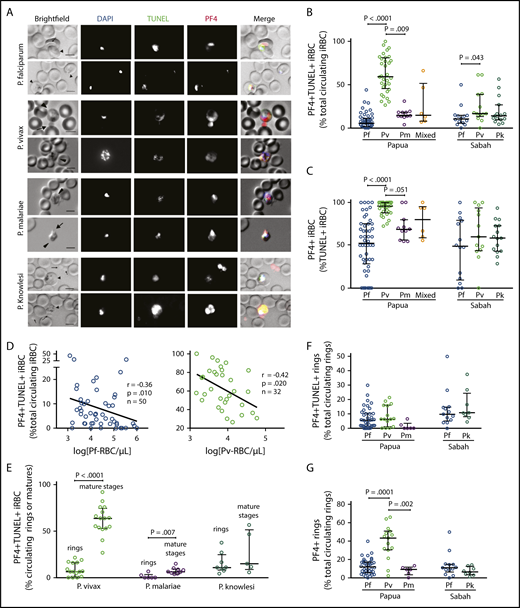
PF4-associated parasite killing in clinical malaria samples. (A) Representative immunofluorescent images from Pf, Pv, Pk, and Pm patient blood smears illustrating PF4-associated parasite killing (PF4+TUNEL+ iRBCs). Scale bars, 5 µm. Arrows and arrowheads indicate platelets and parasites, respectively. Images were taken at 630× magnification on an Axio Scope A1 fluorescent microscope coupled to an Axiocam ICm-1 CCD camera, or an Axio Observer inverted fluorescence microscope coupled to an Axiocam 503 monochrome camera. ZEN 2 software was used for image acquisition and processing (all from Carl Zeiss, Germany). (B) Percentage of PF4+TUNEL+ parasites in clinical samples with Pf (Papua, n = 50; Sabah, n = 14), Pv (Papua, n = 32; Sabah, n = 13), Pm (n = 11), Pk (n = 15), and mixed species infection (n = 6). (C) Comparison of intraerythrocytic PF4 (PF4+) parasites as a percentage of dying (TUNEL+) parasites in Pf, Pv, Pm, Pk, and mixed species infection from Papua and Sabah (n as per B). (D) Inverse correlation of PF4+TUNEL+ iRBCs with parasitemia in Pf and Pv clinical samples (Spearman). (E) Proportions of PF4+TUNEL+ rings vs mature stages in Pv (rings, n = 15; mature, n = 16), Pm (rings, n = 6; mature, n = 9), and Pk (rings, n = 8; mature, n = 5). (F) Proportions of rings that were PF4+TUNEL+ and (G) PF4+ in Pf (Papua, n = 50; Sabah, n = 14), Pv (n = 15), Pm (n = 6), and Pk clinical samples (n = 8). Scatterplots indicate median ± interquartile range for each group. Parasitemia values are log transformed. Kruskal-Wallis or Mann-Whitney test used for statistical comparisons. Data presented in Table 4.
PF4-associated parasite killing in clinical malaria samples. (A) Representative immunofluorescent images from Pf, Pv, Pk, and Pm patient blood smears illustrating PF4-associated parasite killing (PF4+TUNEL+ iRBCs). Scale bars, 5 µm. Arrows and arrowheads indicate platelets and parasites, respectively. Images were taken at 630× magnification on an Axio Scope A1 fluorescent microscope coupled to an Axiocam ICm-1 CCD camera, or an Axio Observer inverted fluorescence microscope coupled to an Axiocam 503 monochrome camera. ZEN 2 software was used for image acquisition and processing (all from Carl Zeiss, Germany). (B) Percentage of PF4+TUNEL+ parasites in clinical samples with Pf (Papua, n = 50; Sabah, n = 14), Pv (Papua, n = 32; Sabah, n = 13), Pm (n = 11), Pk (n = 15), and mixed species infection (n = 6). (C) Comparison of intraerythrocytic PF4 (PF4+) parasites as a percentage of dying (TUNEL+) parasites in Pf, Pv, Pm, Pk, and mixed species infection from Papua and Sabah (n as per B). (D) Inverse correlation of PF4+TUNEL+ iRBCs with parasitemia in Pf and Pv clinical samples (Spearman). (E) Proportions of PF4+TUNEL+ rings vs mature stages in Pv (rings, n = 15; mature, n = 16), Pm (rings, n = 6; mature, n = 9), and Pk (rings, n = 8; mature, n = 5). (F) Proportions of rings that were PF4+TUNEL+ and (G) PF4+ in Pf (Papua, n = 50; Sabah, n = 14), Pv (n = 15), Pm (n = 6), and Pk clinical samples (n = 8). Scatterplots indicate median ± interquartile range for each group. Parasitemia values are log transformed. Kruskal-Wallis or Mann-Whitney test used for statistical comparisons. Data presented in Table 4.
Platelet-associated parasite killing (TUNEL+PF4+ iRBCs) between species and stages
| Patient cohort and Plasmodium species | Samples analyzed, n | Median number parasites counted per sample (IQR) | Median % TUNEL+PF4+ iRBCs (IQR) | P* | P† |
|---|---|---|---|---|---|
| Papua | |||||
| P falciparum | 50 | 54 (52-59) | 5.6 (1.8-11.3) | <.0001 | — |
| P vivax | 32 | 54 (51-58) | 59.3 (45.4-81.1) | Comparator | — |
| P malariae | 11 | 45 (39-50) | 14.3 (13.6-18.2) | .009 | — |
| Mixed infection | 6 | 59 (42-66) | 14.9 (7.8-51.5) | .150 | — |
| P vivax (ring) | 15 | 48 (28-61) | 6.4 (1.1-16.1) | — | Comparator |
| P vivax (mature) | 16 | 59 (44-88) | 63.6 (52.9-74.1) | — | <.0001 |
| P malariae (ring) | 6 | 12 (16-18) | 0 (0-3.5) | — | Comparator |
| P malariae (mature) | 9 | 40 (63-80) | 6.3 (4.4-8.8) | — | .007 |
| Sabah | |||||
| P falciparum | 14 | 95 (76-131) | 10.6 (5.4-14.6) | .043 | — |
| P vivax | 13 | 49 (21-104) | 16.7 (13.5-39.1) | Comparator | — |
| P knowlesi | 15 | 95 (78-101) | 14.2 (19.8-26.9) | .717 | — |
| P knowlesi (ring) | 8 | 96 (92-103) | 10.8 (8.0-24.3) | — | Comparator |
| P knowlesi (mature) | 5 | 78 (72-98) | 14.9 (8.6-51.5) | — | .354 |
| Patient cohort and Plasmodium species | Samples analyzed, n | Median number parasites counted per sample (IQR) | Median % TUNEL+PF4+ iRBCs (IQR) | P* | P† |
|---|---|---|---|---|---|
| Papua | |||||
| P falciparum | 50 | 54 (52-59) | 5.6 (1.8-11.3) | <.0001 | — |
| P vivax | 32 | 54 (51-58) | 59.3 (45.4-81.1) | Comparator | — |
| P malariae | 11 | 45 (39-50) | 14.3 (13.6-18.2) | .009 | — |
| Mixed infection | 6 | 59 (42-66) | 14.9 (7.8-51.5) | .150 | — |
| P vivax (ring) | 15 | 48 (28-61) | 6.4 (1.1-16.1) | — | Comparator |
| P vivax (mature) | 16 | 59 (44-88) | 63.6 (52.9-74.1) | — | <.0001 |
| P malariae (ring) | 6 | 12 (16-18) | 0 (0-3.5) | — | Comparator |
| P malariae (mature) | 9 | 40 (63-80) | 6.3 (4.4-8.8) | — | .007 |
| Sabah | |||||
| P falciparum | 14 | 95 (76-131) | 10.6 (5.4-14.6) | .043 | — |
| P vivax | 13 | 49 (21-104) | 16.7 (13.5-39.1) | Comparator | — |
| P knowlesi | 15 | 95 (78-101) | 14.2 (19.8-26.9) | .717 | — |
| P knowlesi (ring) | 8 | 96 (92-103) | 10.8 (8.0-24.3) | — | Comparator |
| P knowlesi (mature) | 5 | 78 (72-98) | 14.9 (8.6-51.5) | — | .354 |
IQR, interquartile range; PF4, platelet factor-4.
P values from Kruskal-Wallis with Dunn’s multiple comparisons test for difference to P vivax.
P values from Mann-Whitney test between intraspecies ring and mature stages.
We compared PF4+TUNEL+ frequencies in young (ring) vs mature (trophozoite and schizont) asexual stage parasites (Figure 2E; Table 4). In the Papuan P vivax samples, PF4+TUNEL+ mature-stage parasites outnumbered ring-stages almost 10 times (63.6% vs 6.4%; P < .0001); there was a similar significant difference in P malariae (6.3% vs 0%; P = .007), but not in P knowlesi (14.9% vs 10.8%; P = .354). Only ring-stage parasites were observed in the P falciparum samples, consistent with the ability of mature stages to sequester in tissues. There were no significant differences in the proportions of PF4+TUNEL+ rings between patient location or parasite species (Figure 2F). All ring-stage parasites (ie, TUNEL+ and TUNEL−) were also categorized according to PF4 staining; the greatest proportions were observed in Papuan P vivax samples (median, 43.8%), which was significantly greater than in P falciparum or P malariae (median, 12.0% and 9.1%, respectively; Figure 2G).
Collectively, these data show that irrespective of species, a substantial proportion of Plasmodium accumulate platelet-derived PF4 and undergo intraerythrocytic death, most likely as a consequence of the known cytotoxic actions of PF4.
Platelet-derived PF4 accumulation in iRBCs and parasite killing are not associated with systemic platelet activation
We observed that not all PF4-stained parasites were bound by platelets (range, 0%-25%), and some platelet-bound cells were not stained for PF4 (range, 0%-5.9%; supplemental Table 2). We determined whether systemic platelet activation contributes to absorption of plasma PF4 into the iRBCs. None of our markers for platelet activation, including plasma PF4 concentrations, platelet surface expression of CD62P (P-selectin), and PAC-1 (activated GPIIb/IIIa) on circulating platelets, were significantly elevated in any of the patient groups (supplemental Table 3). In addition, no relationships were observed that supported a role for systemic platelet activation in the iRBC accumulation of PF4 (supplemental Table 4).
Cultured P knowlesi and P falciparum are sensitive to human platelets and PF4
In vitro platelet killing of P falciparum has been demonstrated previously,12,29-31 but not for other human Plasmodium species. We therefore cocultured P knowlesi with or without different concentrations of washed and rested human platelets and measured effects on parasite growth over the course of 24 hours. We observed a significant and platelet concentration-dependent reduction in parasite growth, maximal at 75 million platelets/mL (∼60% reduction; Figure 3A). Implementation of platelet-to-iRBC ratios and parasitemias comparable to clinical settings, as well as strict control of platelet quality (supplemental Figure 4), were critical components of our experimental system. A similar significant growth inhibition effect was also observed in P falciparum cultures treated with the same platelet preparations (Figure 3B). P knowlesi was sensitive to recombinant human PF4 (IC50 ∼0.5 µM) and to treatment with platelet lysates, which was blocked by inclusion of anti-PF4 antibodies (Figure 3C-D). Platelet inhibition of parasite growth was prevented when platelets and P knowlesi-iRBCs were physically separated in cocultures using cell-impermeable transwells, but remained sensitive to platelet lysate and PF4 (Figure 3E), indicating that direct platelet–cell contact and platelet-derived PF4 are responsible for the cytotoxic effects.
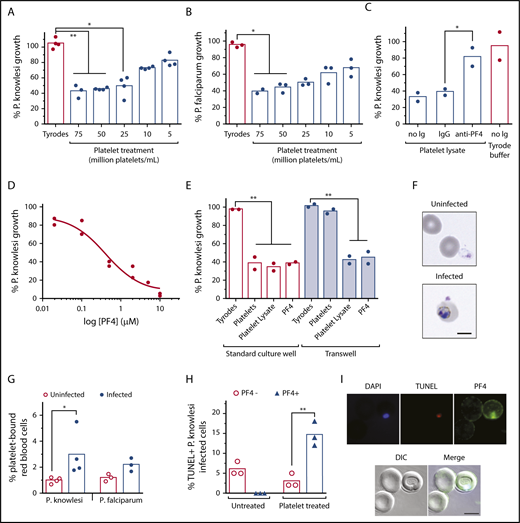
In vitro cultures of P knowlesi are sensitive to platelets and PF4. The growth of (A) P knowlesi (n = 4) and (B) P falciparum (n = 3) treated with different platelet concentrations or Tyrodes buffer for 48 hours. (C) The growth of P knowlesi treated with platelet lysate, with and without anti-PF4 antibodies or immunoglobulin G isotype control (n = 2). (D) P knowlesi PF4 dose-response curve (n = 2). (E) The growth of P knowlesi treated with platelets (60 million/mL), platelet lysate or PF4 (0.5 µM), and cocultured in standard wells or Transwells (n = 2). (F) Micrographs showing platelets bound to uninfected and P knowlesi-infected cells. (G) Percentage platelet binding to uninfected, P knowlesi (n = 4) or P falciparum iRBCs (n = 3), determined by flow cytometry. (H) Percentage TUNEL-labeled (TUNEL+) P knowlesi parasites costained for PF4 (PF4+) or not PF4-stained (PF4−; n = 3). (I) Micrographs showing a PF4+TUNEL+P knowlesi infected cell after platelet treatment. Scale bars, 5 µm. Images were taken at 630× magnification on an Axio Observer inverted fluorescence microscope coupled to an Axiocam 503 monochrome camera with ZEN 2 software (Carl Zeiss, Germany). Bars indicate means of replicate data points. Kruskal-Wallis test or 1-way ANOVA used for statistical comparisons, *P < .05 and **P < .01. DIC, differential interference contrast.
In vitro cultures of P knowlesi are sensitive to platelets and PF4. The growth of (A) P knowlesi (n = 4) and (B) P falciparum (n = 3) treated with different platelet concentrations or Tyrodes buffer for 48 hours. (C) The growth of P knowlesi treated with platelet lysate, with and without anti-PF4 antibodies or immunoglobulin G isotype control (n = 2). (D) P knowlesi PF4 dose-response curve (n = 2). (E) The growth of P knowlesi treated with platelets (60 million/mL), platelet lysate or PF4 (0.5 µM), and cocultured in standard wells or Transwells (n = 2). (F) Micrographs showing platelets bound to uninfected and P knowlesi-infected cells. (G) Percentage platelet binding to uninfected, P knowlesi (n = 4) or P falciparum iRBCs (n = 3), determined by flow cytometry. (H) Percentage TUNEL-labeled (TUNEL+) P knowlesi parasites costained for PF4 (PF4+) or not PF4-stained (PF4−; n = 3). (I) Micrographs showing a PF4+TUNEL+P knowlesi infected cell after platelet treatment. Scale bars, 5 µm. Images were taken at 630× magnification on an Axio Observer inverted fluorescence microscope coupled to an Axiocam 503 monochrome camera with ZEN 2 software (Carl Zeiss, Germany). Bars indicate means of replicate data points. Kruskal-Wallis test or 1-way ANOVA used for statistical comparisons, *P < .05 and **P < .01. DIC, differential interference contrast.
Platelets were observed physically bound to P knowlesi-iRBCs (Figure 3F). The frequency of platelet–iRBC complexes was twice of platelet–uRBC complexes in both P knowlesi and P falciparum after 24 hours of incubation with platelets and was significant for P knowlesi (P < .05; Figure 3G). Platelet-treated P knowlesi cultures contained significantly greater proportions of TUNEL+ iRBCs; the majority of these iRBCs were also PF4+ (Figure 3H-I). PF4 was not detected in untreated iRBCs (Figure 3H). Collectively, human platelets can kill both P falciparum and P knowlesi asexual blood-stage parasites under well-defined culture conditions. The killing mechanism requires platelet–cell contact and PF4; intraerythrocytic PF4 is cytotoxic to P knowlesi.
Discussion
Here we demonstrate for the first time in humans that platelets can directly kill a microbial pathogen, erythrocytic stage Plasmodium. Platelets were observed bound to Plasmodium-iRBCs of all 4 of the major species that cause human disease: P falciparum, P vivax, P malariae, and P knowlesi. Platelet binding to uRBCs was also observed, although the proportions of these were significantly lower than platelet–iRBC complexes. In addition, the proportions of platelet–iRBC complexes were inversely related to parasite burden, suggesting a potential cause and effect relationship between cell–cell binding and parasite growth. We observed frequent intracellular accumulation of PF4 in these iRBCs and death of the intraerythrocytic parasites. Further evidence of platelet’s cytotoxic mechanism of action against parasites was obtained using cultures of P knowlesi and P falciparum. The frequent occurrence of platelet-associated killing of parasites in the periphery of patients with malaria, and its inverse correlation with parasite load, significant at least for P falciparum and P vivax, suggests the importance of platelets in the host control of parasites. Collectively, our data predict that platelets may kill as many as 5% to 20% of circulating blood-stage Plasmodium in clinical malaria, and in P vivax this percentage may be as high as 60%.
We have previously proposed a mechanism of PF4 accumulation and parasite killing involving direct platelet–iRBC contact followed by local release of PF4 and uptake into the parasite via the Duffy antigen.30 Here, our in vitro studies using transwell filters to separate platelets and P knowlesi parasites demonstrated that direct platelet–cell contact is necessary for parasite killing, which confirms previous studies.31 Our cumulative in vitro data contrast with recent findings reported by Gramaglia and colleagues,32 who found no parasite growth inhibition by platelets in P falciparum cultures. We predict this is a result of differences in experimental design, noting especially that in vitro parasite killing by platelets is only observed in cultures containing physiologically relevant proportions of parasite-infected cells (<1%, in this study and previously12,31 ). The aforementioned study also did not observe changes in parasite growth rates when platelet levels were altered in Plasmodium-infected mice or evidence that platelets were required for survival, which conflicts with the findings of others.12,13 This could be a result of differences in the Plasmodium strains used among the studies, which can affect parasite virulence, growth, and sequestration and host response characteristics. In addition, recognizing the definitive parasite killing activity of platelets in the circulation requires distinguishing dead parasites from healthy parasites, which are greatest in platelet-sufficient mice12 and in parasitized cells that contain platelet-derived PF4 (this study and McMorran et al30 ). In the patient studies, we also considered and excluded the possibility that PF4 is absorbed by circulating parasites from the plasma. Systemic platelet activation was negligible, and plasma PF4 concentrations were approximately 1000-fold lower than in vitro concentrations required for Plasmodium killing. We also observed substantially higher frequencies of PF4 accumulation in mature-stage P vivax-iRBCs and greater rates of parasite death. Accumulation may be determined by the higher Duffy antigen levels expressed on reticulocytes,42-44 for which P vivax has an exclusive tropism.45 Although circulating P vivax parasitemia is intrinsically limited by the number of circulating reticulocytes, we speculate that the greater degree of platelet-associated killing observed in P vivax infection may be a substantial additional contributor to the lower parasitemia generally observed in these patients compared with P falciparum.18,46,47
Platelet cytoadherence to the endothelium and WBCs are well-established pathological drivers of vascular and inflammatory diseases. However, the occurrence and consequences of platelets interacting with RBCs in any disease setting have been underreported.48-50 Our unique ability to systematically characterize platelet–RBC interactions revealed that platelets have a greater capacity to bind Plasmodium-iRBCs vs uRBCs. This may be partly determined by parasite-expressed proteins present on the RBC surface, such as the P falciparum cell adhesion molecule PfEMP1 shown previously to mediate platelet binding through CD36.51 Platelet binding to uRBC may be mediated through erythroid-expressed ICAM-4 and platelet GPIIb/IIIa.52 The identities and roles of other RBCs and platelet molecules involved in platelet–RBC binding, especially in the other Plasmodium species, remain to be determined.
Platelet–RBC complexes may have a role in malaria-induced thrombocytopenia, which has been reported in many other clinical studies53-55 and was also evident in our patient groups. The underlying causes of thrombocytopenia have been variously attributed to systemic platelet activation, immune-mediated clearance, and vascular pooling.56-61 Platelets complexed with RBCs are not recognized by hematological analyzers; thus, complex formation would lead to an apparent platelet loss. The high frequencies of these complexes relative to free platelets in our patients suggest they make up a substantial proportion of the total platelet pool. If there is an accelerated turnover of these complexes, this would further enhance the contribution of complexes to platelet loss. Interestingly, host mechanisms that remove diseased and damaged circulating cells such as the spleen are upregulated in malaria62 ; the lifetime of platelet–RBC complexes in the circulation remains unknown. Further supporting this hypothesis, platelet–RBC complexes were greatest in our knowlesi patients with malaria, and this species causes the highest frequency of thrombocytopenia.35,53
Platelet activation and binding is considered a key mechanism in enabling sequestration of P falciparum-iRBCs to microvasculature, and platelet-mediated iRBC sequestration is associated with fatal outcome in cerebral malaria studies.63-66 Our data in predominantly nonsevere P falciparum, P vivax, and P malariae indicated negligible systemic platelet activation. Systemic platelet activation in severe and nonsevere falciparum malaria were comparable; however, numbers were small and we cannot exclude greater systemic platelet activation in human severe malaria,67,68 nor can we exclude tissue platelet activation not detected by the circulating measures used in this study.
Overall, our study demonstrates the direct pathogen-killing actions and host protective roles of platelets during human malarial infection. Given platelets show broad-spectrum antimicrobial activity, and the risks for infection associated with thrombocytopenia and platelet disorders are elevated, a general role for platelets in the innate host defense against microbial infection should be considered.
Presented as an oral presentation at the 6th International Conference on Plasmodium vivax Research, Manaus, Brazil, 14 June 2017.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Pak Prayoga, Leo Leonardo, and Ruland Wandosa for their microscopy expertise; Bernadette Pedersen, Timothy Butler, Harpret Vohra, and Michael Devoy for flow cytometry assistance; Guy Zimmerman for advice on measures of platelet activation; Grennady Wirjanata for assistance in the Timika laboratory; and Yati Soenarto for facilitating the Papua study. The authors thank study participants; Mitra Masyarakat Hospital Staff in Papua; the malaria research team, nursing and laboratory staff at Queen Elizabeth, Kudat, Kota Marudu, and Pitas Hospitals in Sabah; and the Director General of Health Malaysia for permission to publish the work.
This work was supported by the Australian National Health and Medical Research Council (Grants #1037304, #1045156, #490037, #605524, #1047082, #1047090, and #1066502, and Fellowships to N.M.A. [#1042072, #1135820], B.E.B. [#1088738], and M.J.G. [#1138860]), the Australian Research Council (grant #120100061), the Wellcome Trust (Fellowships to R.N.P. [#200909] and J.R.P. [#099875]), the Singapore National Medical Research Council (Award to T.W.Y. [CSA INV 15nov007]), and the Australian Department of Foreign Affairs and Trade.
Funders had no role in the design of the study and collection, analysis, and interpretation of data or in writing the manuscript.
Authorship
Contribution: S.K., B.E.B., T. Woodberry, S.F., G.M., T.W.Y., N.M.A., and B.J.M. provided conceptualization; S.K., B.E.B., E.J., K.A.P, A.E., T. Woodberry, G.M., M.J.G., N.M.A., and B.J.M. provided methodology; S.K., B.E.B., E.J., K.A.P., A.E., and B.J.M. provided validation; S.K., B.E.B., E.J., M.J.G., and B.J.M. provided formal analysis; S.K., B.E.B., E.J., B.A., E.K., K.A.P., A.E., T. Williams, M.J.G., N.M.A., and B.J.M. provided investigation; S.K., B.E.B., J.R.P., E.K., K.A.P., R.N.P., T. Williams, S.F., N.M.A., and B.J.M. provided resources; S.K., B.E.B., M.J.G., N.M.A., and B.J.M. wrote the original draft; J.R.P., E.K., R.N.P., T. Woodberry, S.F., G.M., and T.W.Y. performed review and editing; S.K. and B.J.M. performed visualization; J.R.P., N.M.A., and B.J.M. provided supervision; S.K., B.E.B., and K.A.P. performed project administration; and R.N.P., N.M.A., and B.J.M. provided funding acquisition.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Brendan J. McMorran, The John Curtin School of Medical Research, Australian National University, Canberra, ACT 2601, Australia; e-mail: brendan.mcmorran@anu.edu.au.
