

Key Points
Somatic mutations driving clonal hematopoiesis occur mainly in DNMT3A and TET2 and have no significant impact on hematological phenotypes.
There is a familial predisposition to acquire TET2 mutation.
Abstract
Age-associated clonal hematopoiesis caused by acquired mutations in myeloid cancer–associated genes is highly prevalent in the normal population. Its etiology, biological impact on hematopoiesis, and oncogenic risk is poorly defined at this time. To gain insight into this phenomenon, we analyzed a cohort of 2530 related and unrelated hematologically normal individuals (ages 55 to 101 years). We used a sensitive gene-targeted deep sequencing approach to gain precision on the exact prevalence of driver mutations and the proportions of affected genes. Mutational status was correlated with biological parameters. We report a higher overall prevalence of driver mutations (13.7%), which occurred mostly (93%) in DNMT3A or TET2 and were highly age-correlated. Mutation in these 2 genes had some distinctive effects on end points. TET2 mutations were more age-dependent, associated with a modest neutropenic effect (9%, P = .012), demonstrated familial aggregation, and associated with chronic obstructive pulmonary disease. Mutations in DNMT3A had no impact on blood counts or indices. Mutational burden of both genes correlated with X-inactivation skewing but no significant association with age-adjusted telomere length reduction was documented. The discordance between the high prevalence of mutations in these 2 genes and their limited biological impact raise the question of the potential role of dysregulated epigenetic modifiers in normal aging hematopoiesis, which may include support to failing hematopoiesis.
Introduction
Aging hematopoiesis is associated with decreased bone marrow cellularity,1 reduced lymphopoiesis,2 and more anemia.3 Older hematopoietic stem cells have defective self-renewal capacity4-6 and become myeloid-biased.7-9 There is an age-related increase in incidence of myeloid cancers such as acute myeloid leukemias (AML), myelodysplastic syndromes (MDSs), and myeloproliferative neoplasms (MPNs) (www.cancer.gov/statistics). Importantly, the development of hematological cancers has recently been linked to age-associated clonal hematopoiesis.10,11
Age-associated clonal hematopoiesis was first suggested by X-chromosome inactivation (XCI) analyses performed in normal aging females more than 20 years ago.12,13 The etiology(ies) of this clonal dominance remained hypothetical until the documentation of acquired TET2 mutations in a subset of these hematologically normal individuals.14 The documentation of acquired clonal mosaicism based on copy number anomalies further supported the age-associated prevalence of clonal hematopoiesis.15-19 In 2014, 3 groups reported analysis of DNA exome datasets10,11,20 and documented age-dependent mutations in genes associated with hematological cancers. Although more than 70 different genes have been identified, the most frequently mutated are DNMT3A, TET2, ASXL1, TP53, JAK2, SF3B1, CBL, SRSF2, PPM1D, and BCOR.10,11,20 Importantly, the prospective data available for 2 of these studies documented a relative risk of 11.111 to 12.910 of developing an hematological cancer in subjects with clonal hematopoiesis. Although the prospective association with hematological cancer has been established, prediction of individual risk remains uncertain. Asymptomatic subjects are considered to have clonal hematopoiesis of indeterminate potential.21
The aim of this study is to increase our understanding of age-associated clonal hematopoiesis by studying a cohort of 2530 related and unrelated individuals. We precisely determined the prevalence of mutations using a high-resolution targeted gene sequencing approach on myeloid cells obtained from fractionated blood specimens. We investigated the biological impact of these mutations by correlation analysis of several parameters including blood counts and indices, telomere length (TL), XCI, and familial aggregation.
Methods
Cohort
The study population comprised 2530 women of French-Canadian ancestry without any known hematological disorder, ranging from 55 to 101 years of age. Family-based subjects were 1727 individuals belonging to 435 sib-ships. There were 803 unrelated subjects. All subjects answered a medical questionnaire and gave informed consent. The study was approved by the Maisonneuve-Rosemont Hospital’s Ethics Committee in 1998 and reapproved annually. Demographic of the cohort is presented in Figure 1A.

Prevalence and distribution of somatic mutations in aging hematopoiesis. (A) Age distribution of the 2530 women of the cohort. (B) Prevalence of somatic mutation in the 347 mutated individuals of the cohort; the pale blue shade represents 95% confidence interval. (C) Distribution of the types of single-nucleotide substitutions observed in somatic variants of the cohort. (D) Allelic fraction distribution of the 402 somatic mutations observed. (E) Contribution of individual genes to the total number of observed somatic mutations. VAF, variant allele fraction.
Prevalence and distribution of somatic mutations in aging hematopoiesis. (A) Age distribution of the 2530 women of the cohort. (B) Prevalence of somatic mutation in the 347 mutated individuals of the cohort; the pale blue shade represents 95% confidence interval. (C) Distribution of the types of single-nucleotide substitutions observed in somatic variants of the cohort. (D) Allelic fraction distribution of the 402 somatic mutations observed. (E) Contribution of individual genes to the total number of observed somatic mutations. VAF, variant allele fraction.
Sample processing, XCI, and telomere length determination
Blood cells were obtained by venipuncture and buccal epithelial cells using a cotton and paper swab (Whatman Bioscience). Complete blood counts were obtained from a GenS automated cell counter (Beckman Coulter). Blood cells were separated into polymorphonuclear (PMN) and mononuclear fractions by standard density gradient centrifugation (Ficoll-Paque) and DNA obtained by standard procedures. XCI was assessed using the HUMARA assay as previously described.22,23 TL in whole blood was measured using the method of Cawthon,24 with slight modification.25
Myeloid gene sequencing and bioinformatics analyses
PMNs were sequenced at high coverage on an Ion Proton sequencer (Thermo Fisher Scientific) using the Ampliseq AML panel covering 19 recurrently mutated genes in myeloid cancers: DNMT3A, TET2, ASXL1, TP53, JAK2, BRAF, CBL, CEBPA, FLT3, GATA2, IDH1, IDH2, KIT, KRAS, NPM1, NRAS, PTPN11, RUNX1, and WT1. This panel covers 90% of the mutations reported, including the 5 most prevalent. We generated 237 amplicons >22 kb. Each sample was sequenced at a minimum of 4000× mean coverage (corresponding to 95% >500×) and aligned to the human reference genome (hg19) using the Torrent Suite software v4.6 (Thermo Fisher Scientific). Mutations were detected using VariantCallerv4.6, then annotated with IonReporterv4.6. Subsequently, mutations were filtered using IonReporter and only exonic and splice site mutations with a minor allele frequency ≥0.02 were kept for further annotation. Frameshift, nonsense, in-frame deletions or insertions, splice sites, and missense mutations with a PolyPhen score >0.99 (or no score) were considered significant. Putative germline mutations were confirmed by sequencing DNA from buccal epithelial cells using the same method. For validation, 10 control samples with known TET2 mutations14 were sequenced on Ion Proton. Additionally, the 31 first TET2 mutations were validated by Sanger resequencing and analytical sensitivity was validated (see supplemental Table 1 and supplemental Figure 1, available on the Blood Web site). SF3B1 (not part of the panel) was sequenced in a cohort of 207 individuals (71 to 98 years of age) using a custom Ampliseq panel covering the entire coding sequence. No somatic mutations were identified and further analysis was abandoned.26
5hmC and 5mC measurement
Genomic DNA from total blood cells was hydrolyzed using DNA Degradase Plus (Zymo Research) following the manufacturer’s recommendations. Reaction was stopped by the addition of formic acid (0.1%) spiked with stable isotope-labeled internal standards. Global 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) levels were assessed by liquid chromatography-electrospray ionization-tandem mass spectrometry with multiple reaction monitoring (LC-ESI-MS/MS-MRM) as described previously.27
Statistical analysis
The descriptive statistics and analyses were performed using SAS 9.4 (SAS Institute Inc., Cary, NC). Outliers were identified and removed based on mean ±4× standard deviation for each end point. Subjects homozygous at the HUMARA locus were eliminated for the XCI analysis. Some telomere data were unavailable. Nine parameters were used as end points: total white blood cells (WBC) (n = 2493), hemoglobin (Hb) (n = 2494), absolute lymphocyte counts (n = 2493), absolute monocyte counts (n = 2495), absolute neutrophil counts (n = 2494), platelets (n = 2488), mean corpuscular volume (n = 2495), clonality (degree of XCI skewing in PMN) (n = 1989) and TL (n = 2006). Importantly, we have identified end point-altering variables in the cohort variables, other than mutations. These included age for XCI, TL, and all hematological parameters. Smoking was identified for WBC, Hb, lymphocytes, neutrophils, and MCV. All analyses examining effects of mutations on end points were controlled for these variables. The covariates of age and smoking were considered as fixed effects and the family was used as random effect in a linear mixed model. Log10-transformation was applied to non-normal end points to achieve normality. Mutated individuals were treated in a binary fashion (0 = no mutation, 1 = mutation) and analyzed separately: (1) any mutation; (2) DNMT3A mutation; (3) TET2 mutation; (4) DNMT3A mutation with VAF ≥10%; and (5) TET2 mutation with VAF ≥10%. The DNMT3A or TET2 VAF was also considered as continuous quantitative trait in the entire cohort. Significance level was fixed at .0024 after Bonferroni correction to account for multiple testing. However, all results showing P ≤ .05 are reported as being of potential interest. To evaluate the genetic risk of acquiring a mutation in DNMT3A or TET2, we used the recurrence-risk ratio of disease in siblings, (λs), calculated using these formulae28 :

Ks recurrence risk, where ns(a) is the number of sib-ship of size s and a affected.

Prevalence in the general population (K) was evaluated in unrelated individuals age-matched with individuals from the pedigrees.
Results
DNMT3A and TET2 somatic mutations account for the vast majority of acquired mutations
Validation of the sequencing approach.
The validation is presented in supplemental Table 1 and supplemental Figures 1 and 2. Importantly, DNMT3A coding sequence (exons 1 to 23) was covered at 100% by 42 amplicons. For TET2, 59 amplicons covering 100% of the coding sequence (exons 3 to 11) were used.
Prevalence of mutations.
A total of 2542 individuals were sequenced. Twelve with germline mutations (5 in DNMT3A, 4 in TET2, 2 in ASXL1, and 1 in GATA2) were excluded from the analysis. In the remaining 2530 individuals, we identified 402 somatic mutations in 347 individuals (13.7%) (Figure 1A-B). Similarly to previous reports,10,11 one-half of the single nucleotide protein identified corresponded to a C>T substitution, a mutational signature characteristic of aging (Figure 1C).29,30 The VAF was measured for all subjects and varied between 3.9% and 91.5% (mean, 14.3%). Only 3 individuals had a somatic mutation with a VAF 50%, suggesting somatic recombination (JAK2 V617F at 60.1%, TET2 Y1245fs at 74.8%, and ASXL1 A640fs at 91.5%). A VAF of ≥10%, corresponding to 20% of mutated cells was present in 46.4% of mutated individuals (Figure 1D). The most commonly affected genes were the epigenetic regulators DNMT3A and TET2, accounting, respectively, for 57.2% (230/402) and 33.3% (134/402) of all documented mutations and representing 92.8% (322/347) of all mutated individuals (Figure 1E). Mutations were also recurrently observed in ASXL1 (n = 10; 2.5%), TP53 (n = 10; 2.5%), JAK2 (n = 9; 2.2%), CBL (n = 3; 0.7%), KRAS (n = 2; 0.5%), NRAS (n = 1; 0.2%), RUNX (n = 1; 0.2%), CEBPA (n = 1; 0.2%), and IDH2 (n = 1; 0.2%) (Figure 1E).
Multiple mutations.
Fifty of the mutated individuals (14.4%) had more than 1 mutation (Figure 2). The majority (46/50) had 2 mutations, 3 had 3 mutations, and 1 had 4 mutations (Figure 2A). The multiple mutant subgroup was significantly older than the single mutant one (75.7 vs 72.8 years of age, P < .05). Furthermore, not only was the maximum VAF higher in these individuals (19.0% vs 13.5%, P < .005), but the VAF in triple or quadruple mutants was higher than those of the single or double mutants (41.4% vs 17.0%, P < .0001), suggesting time-dependent clonal evolution. The most frequent combination (23/50) was a second mutation in the same gene (double DNMT3A or double TET2) followed by combined DNMT3A and TET2 mutations (12/50) (Figure 2B). When we evaluated the respective VAF between mutated genes to identify the sequence of events, no systematic precedence was observed in subjects with mutation in both DNMT3A and TET2.

Cooccurring mutations distribution and VAF. (A) Cooccurrence of the 402 somatic mutations observed in the 347 mutated individuals of the cohort. Darker shade represents double mutation in the same gene. (B) VAF of all 50 individuals with cooccurring mutations including individuals harboring double mutation in TET2 or DNMT3A.
Cooccurring mutations distribution and VAF. (A) Cooccurrence of the 402 somatic mutations observed in the 347 mutated individuals of the cohort. Darker shade represents double mutation in the same gene. (B) VAF of all 50 individuals with cooccurring mutations including individuals harboring double mutation in TET2 or DNMT3A.
Type of mutations occurring in DNMTA3 and TET2.
Mutations in DNMT3A and TET2 were spread over the entire coding sequence, with missense mutations clustering in known structural and functional domains (Figure 3). For DNMT3A, 230 somatic mutations were found, including 27 indels, 30 nonsenses, 31 splice sites, and 142 missenses (21/142 at position R882) (Figure 3A). For TET2, 134 somatic mutations were identified: 50 indels, 35 nonsenses, 6 splice sites, and 48 missenses (Figure 3B).

Schematic diagram of DNMT3A and TET2 mutations. (A) Distribution of the 230 somatic mutations observed in DNMT3A. (B) Distribution of the 134 somatic mutations observed in TET2. Blue dots, missense mutations; red dots, truncating mutations (frame shift, nonsense and splice site); gray dots, in-frame deletions or insertions; purple, residues that are affected by different mutation types at the same proportion. The height of the line is proportional to the number of observations (from 1 to 21).
Schematic diagram of DNMT3A and TET2 mutations. (A) Distribution of the 230 somatic mutations observed in DNMT3A. (B) Distribution of the 134 somatic mutations observed in TET2. Blue dots, missense mutations; red dots, truncating mutations (frame shift, nonsense and splice site); gray dots, in-frame deletions or insertions; purple, residues that are affected by different mutation types at the same proportion. The height of the line is proportional to the number of observations (from 1 to 21).
Effect of TET2 mutation on epigenetic marks.
Global 5hmC and 5mC levels were quantified by LC-ESI-MS/MS-MRM.27 We analyzed 465 individuals from the cohort and identified 45 TET2 mutations (15 indels, 11 nonsenses, 17 missenses, and 2 splice sites) in 41 individuals (37 single TET2 mutants and 4 double TET2 mutants). TET2 mutations were associated with significant reductions in 5hmC/5mC ratio compared with aged-matched WT individuals. Moreover, this reduction was proportional to the VAF (Pearson correlation, R2 = 0.8512, P < .0001, Figure 4). This clearly indicates that mutations occurring in aging individuals have a biological impact on epigenome.

Correlation between TET2 VAF and 5hmC/5mC ratio. Global levels of 5hmC and 5mC was quantified by LC-ESI-MS/MS-MRM in a cohort of 465 individual including 41 TET2 mutants. *For double TET2 mutants (n = 4) the sum of the VAF vas used.
Correlation between TET2 VAF and 5hmC/5mC ratio. Global levels of 5hmC and 5mC was quantified by LC-ESI-MS/MS-MRM in a cohort of 465 individual including 41 TET2 mutants. *For double TET2 mutants (n = 4) the sum of the VAF vas used.
Risk factors for acquiring mutations
Age effect on mutation prevalence.
The prevalence of mutations was strongly associated with age: 6.3% in individuals 55 to 59, 11.2% in those 60 to 69, 14.8% in those 70 to 79, 24.5% in those 80 to 89, and 42.9% in individuals older than 90 years of age (Figure 1A-B). These prevalences are twice those reported by Jaiswal et al,11 and 3 to 6 times those documented by Xie et al.20 When considering the mutations as a categorical end point (ie, mutated or not), each additional year was associated with a 1.404 (P < .0001) and 1.0937 (P < .0001) odds ratios of acquiring a mutation in DNMT3A and TET2, respectively (Figure 5A). The difference in age effect between DNMT3A and TET2 was significant (P < .0001). The greater age effect of TET2 explains why DNMT3A is globally the most frequently mutated gene, whereas after 85 years of age, TET2 predominates (Figure 5B).

Prevalence of DNMT3A and TET2 somatic mutation in the cohort in function of age. (A) When considering the mutations as a categorical end point (0/1), effect of 1 year increase in age on the increase in the odds of having a mutation in DNMT3A and TET2. (B) Age of the individuals harboring a DNMT3A (red, n = 215) or TET2 (blue, n = 122) somatic mutations in the cohort. The pale shading represents 95% confidence interval;  , standardized regression coefficient; StdErr., standard error.
, standardized regression coefficient; StdErr., standard error.
Prevalence of DNMT3A and TET2 somatic mutation in the cohort in function of age. (A) When considering the mutations as a categorical end point (0/1), effect of 1 year increase in age on the increase in the odds of having a mutation in DNMT3A and TET2. (B) Age of the individuals harboring a DNMT3A (red, n = 215) or TET2 (blue, n = 122) somatic mutations in the cohort. The pale shading represents 95% confidence interval; , standardized regression coefficient; StdErr., standard error.
Comorbidities associated with mutations.
Using negative binomial regression, we correlated mutation status with self-reported comorbidities including allergies, cardiovascular disease, chronic obstructive pulmonary disease (COPD)-asthma, arthritis, diabetes, hypertension, thyroid disorders, hay fever, cancer, and smoking status. We documented a significant association only between COPD-asthma and TET2 (supplemental Table 2).
Heritability of mutation.
That part of the cohort was recruited in sib-ship allowed us to evaluate for the first time the heritability of mutations. We chose the recurrence-risk ratio of disease in siblings (λs), a standard parameter, to estimate the statistical power for detection of a disease locus.28 Of the 391 sib-ships available, 56 were analyzed for women age ≥55 years of age for TET2 and 98 for DNMT3A (42 and 58 for females age ≥ 65 years of age, respectively) and showed that the recurrent-risk of acquiring a DNMT3A mutation is low to nonexistent. On the other hand, for TET2 mutation, this risk was much higher: 2.24 and 2.65 in individuals more than 55 and 65 years of age, respectively (Figure 6A). This establishes familial aggregation for TET2 acquisition. One sib-ship was particularly informative, with 4/7 having acquired a different TET2 mutation, 1/7 a DNMT3A mutation, and 2 having remained WT (Figure 6B).

Heritability of mutation. (A) Sibling recurrence risk estimation for DNMT3A and TET2 using unrelated ages-matched as controls. (B) Example of a familial cluster of acquired mutations in TET2. The pedigree shows a sib-ship of 7, with 4 harboring different TET2 somatic mutations and 1 a DNMT3A. *, nonsense mutation; fs, frameshift mutation; Ks, sibling recurrence risk; λs, recurrence-risk ratio of disease in siblings.
Heritability of mutation. (A) Sibling recurrence risk estimation for DNMT3A and TET2 using unrelated ages-matched as controls. (B) Example of a familial cluster of acquired mutations in TET2. The pedigree shows a sib-ship of 7, with 4 harboring different TET2 somatic mutations and 1 a DNMT3A. *, nonsense mutation; fs, frameshift mutation; Ks, sibling recurrence risk; λs, recurrence-risk ratio of disease in siblings.
Effect of DNMT3A and TET2 somatic mutations on clonal expansion
Limited with the absence of serial resampling, we estimated whether DNMT3A and TET2 mutations offer a proliferative advantage to cells by examining the relationship between age and VAF in mutated individuals. For each increased year, there was a 3.95% (P < .0001) and 9.98% (P < .0001) increase in the estimated mean VAF of DNMT3A and TET2, respectively. This suggests that clones are not stable but steadily increase and that TET2 mutations may have a stronger effect on proliferation than DNMT3A mutations.
We also evaluated the impact of mutational status on age-adjusted TL (Table 1). TL reduction would support a high proliferative rate, as has already been demonstrated in MPN.31 Analysis was performed for DNMT3A and TET2 independently and for all mutated individuals. Although there was a trend toward shorter TL, no analysis reached significance threshold (Table 1).
Impact of DNMT3A, TET2, or any mutation on blood formula and biological parameters
| End point | Covariates | Trans. | Mutation | 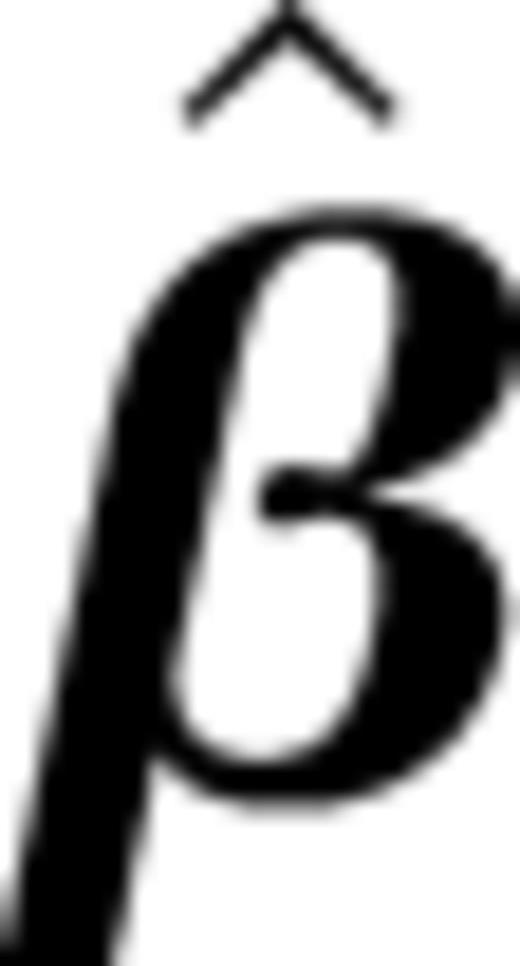 | SE | P | % Change |
|---|---|---|---|---|---|---|---|
| WBC | Age + smoking | log10 | DNMT3A | 0.0142 | 0.0074 | .055 | 3.33 |
| DNMT3A >10% | 0.0123 | 0.0111 | .269 | 2.87 | |||
| DNMT3A VAF continuous | 0.0006 | 0.0004 | .186 | 0.13 | |||
| TET2 | −0.0178 | 0.0098 | .070 | −4.02 | |||
| TET2 >10% | −0.0265 | 0.0128 | .039 | −5.92 | |||
| TET2 VAF continuous | −0.0005 | 0.0005 | .276 | −0.11 | |||
| Any mutation | 0.0035 | 0.0061 | .565 | 0.81 | |||
| Any mutation >10% | −0.0009 | 0.0084 | .910 | −0.22 | |||
| Hemoglobin | Age + smoking | None | DNMT3A | 0.3321 | 0.6645 | .617 | 0.25 |
| DNMT3A >10% | 1.8138 | 0.9933 | .068 | 1.36 | |||
| DNMT3A VAF continuous | 0.0624 | 0.0390 | .110 | NA | |||
| TET2 | −0.3903 | 0.8890 | .661 | −0.29 | |||
| TET2 >10% | −0.9667 | 1.1561 | .403 | −0.73 | |||
| TET2 VAF continuous | −0.0476 | 0.0411 | .247 | NA | |||
| Any mutation | 0.6235 | 0.5467 | .254 | 0.47 | |||
| Any mutation >10% | 1.1440 | 0.7468 | .126 | 0.86 | |||
| Lymphocytes | Age + smoking | log10 | DNMT3A | 0.0085 | 0.0095 | .371 | 1.97 |
| DNMT3A >10% | 0.0099 | 0.0142 | .486 | 2.30 | |||
| DNMT3A VAF continuous | 0.0004 | 0.0006 | .475 | 0.09 | |||
| TET2 | −0.0038 | 0.0127 | .763 | −0.88 | |||
| TET2 >10% | −0.0112 | 0.0165 | .498 | −2.54 | |||
| TET2 VAF continuous | −0.0001 | 0.0006 | .800 | −0.03 | |||
| Any mutation | 0.0071 | 0.0078 | .364 | 1.65 | |||
| Any mutation >10% | 0.0015 | 0.0107 | .885 | 0.35 | |||
| Monocytes | Age + smoking | log10 | DNMT3A | 0.0061 | 0.0098 | .535 | 1.42 |
| DNMT3A >10% | 0.0068 | 0.0148 | .644 | 1.59 | |||
| DNMT3A VAF continuous | 0.0003 | 0.0006 | .630 | 0.07 | |||
| TET2 | 0.0067 | 0.0132 | .614 | 1.54 | |||
| TET2 >10% | −0.0038 | 0.0172 | .824 | −0.88 | |||
| TET2 VAF continuous | 0.0015 | 0.0006 | .013 | 0.35 | |||
| Any mutation | 0.0068 | 0.0082 | .405 | 1.58 | |||
| Any mutation >10% | 0.0038 | 0.0112 | .737 | 0.87 | |||
| Neutrophils | Age + smoking | log10 | DNMT3A | 0.0183 | 0.0096 | .057 | 4.30 |
| DNMT3A >10% | 0.0122 | 0.0144 | .397 | 2.84 | |||
| DNMT3A VAF continuous | 0.0006 | 0.0006 | .294 | 0.14 | |||
| TET2 | −0.0303 | 0.0127 | .017 | −6.73 | |||
| TET2 >10% | −0.0417 | 0.0166 | .012 | −9.15 | |||
| TET2 VAF continuous | −0.0011 | 0.0006 | .053 | −0.26 | |||
| Any mutation | 0.0007 | 0.0079 | .929 | 0.16 | |||
| Any mutation >10% | −0.0058 | 0.0108 | .595 | −1.32 | |||
| Platelets | Age | log10 | DNMT3A | 0.0014 | 0.0068 | .831 | 0.33 |
| DNMT3A >10% | −0.0022 | 0.0102 | .827 | −0.51 | |||
| DNMT3A VAF continuous | −0.0002 | 0.0004 | .702 | −0.03 | |||
| TET2 | −0.0124 | 0.0092 | .175 | −2.82 | |||
| TET2 >10% | −0.0256 | 0.0120 | .033 | −5.73 | |||
| TET2 VAF continuous | −0.0008 | 0.0005 | .073 | −0.19 | |||
| Any mutation | 0.0032 | 0.0056 | .572 | 0.73 | |||
| Any mutation >10% | −0.0017 | 0.0077 | .829 | −0.38 | |||
| MCV | Age + smoking | none | DNMT3A | −0.0710 | 0.2686 | .791 | −0.08 |
| DNMT3A >10% | −0.4800 | 0.4034 | .234 | −0.52 | |||
| DNMT3A VAF continuous | −0.0118 | 0.0157 | .455 | NA | |||
| TET2 | −0.3377 | 0.3535 | .340 | −0.37 | |||
| TET2 >10% | −0.7134 | 0.4616 | .123 | −0.78 | |||
| TET2 VAF continuous | −0.0162 | 0.0163 | .320 | NA | |||
| Any mutation | −0.0263 | 0.2192 | .905 | −0.03 | |||
| Any mutation > 10% | −0.3206 | 0.3011 | .287 | −0.35 | |||
| XCI ratio skewing | Age | none | DNMT3A | 0.0227 | 0.0114 | .046 | 9.56 |
| DNMT3A >10% | 0.0707 | 0.0169 | 3.10E-05 | 29.84 | |||
| DNMT3A VAF continuous | 0.0026 | 0.0006 | 5.67E-05 | NA | |||
| TET2 | 0.0697 | 0.0144 | 1.55E-06 | 29.38 | |||
| TET2 >10% | 0.0924 | 0.0187 | 9.55E-07 | 38.94 | |||
| TET2 VAF continuous | 0.0034 | 0.0007 | 4.20E-07 | NA | |||
| Any mutation | 0.0375 | 0.0092 | 4.76E-05 | 15.80 | |||
| Any mutation >10% | 0.0746 | 0.0124 | 2.53E-09 | 31.42 | |||
| Telomere length | Age | log10 | DNMT3A | −0.0104 | 0.0096 | .279 | −2.37 |
| DNMT3A >10% | −0.0155 | 0.0147 | .292 | −3.51 | |||
| DNMT3A VAF continuous | −0.0008 | 0.0006 | .143 | −0.19 | |||
| TET2 | −0.0254 | 0.0129 | .049 | −5.68 | |||
| TET2 >10% | −0.0073 | 0.0172 | .673 | −1.66 | |||
| TET2 VAF continuous | −0.0010 | 0.0007 | .129 | −0.23 | |||
| Any mutation | −0.0205 | 0.0079 | .010 | −4.60 | |||
| Any mutation >10% | −0.0196 | 0.0110 | .076 | −4.40 |
| End point | Covariates | Trans. | Mutation | | SE | P | % Change |
|---|---|---|---|---|---|---|---|
| WBC | Age + smoking | log10 | DNMT3A | 0.0142 | 0.0074 | .055 | 3.33 |
| DNMT3A >10% | 0.0123 | 0.0111 | .269 | 2.87 | |||
| DNMT3A VAF continuous | 0.0006 | 0.0004 | .186 | 0.13 | |||
| TET2 | −0.0178 | 0.0098 | .070 | −4.02 | |||
| TET2 >10% | −0.0265 | 0.0128 | .039 | −5.92 | |||
| TET2 VAF continuous | −0.0005 | 0.0005 | .276 | −0.11 | |||
| Any mutation | 0.0035 | 0.0061 | .565 | 0.81 | |||
| Any mutation >10% | −0.0009 | 0.0084 | .910 | −0.22 | |||
| Hemoglobin | Age + smoking | None | DNMT3A | 0.3321 | 0.6645 | .617 | 0.25 |
| DNMT3A >10% | 1.8138 | 0.9933 | .068 | 1.36 | |||
| DNMT3A VAF continuous | 0.0624 | 0.0390 | .110 | NA | |||
| TET2 | −0.3903 | 0.8890 | .661 | −0.29 | |||
| TET2 >10% | −0.9667 | 1.1561 | .403 | −0.73 | |||
| TET2 VAF continuous | −0.0476 | 0.0411 | .247 | NA | |||
| Any mutation | 0.6235 | 0.5467 | .254 | 0.47 | |||
| Any mutation >10% | 1.1440 | 0.7468 | .126 | 0.86 | |||
| Lymphocytes | Age + smoking | log10 | DNMT3A | 0.0085 | 0.0095 | .371 | 1.97 |
| DNMT3A >10% | 0.0099 | 0.0142 | .486 | 2.30 | |||
| DNMT3A VAF continuous | 0.0004 | 0.0006 | .475 | 0.09 | |||
| TET2 | −0.0038 | 0.0127 | .763 | −0.88 | |||
| TET2 >10% | −0.0112 | 0.0165 | .498 | −2.54 | |||
| TET2 VAF continuous | −0.0001 | 0.0006 | .800 | −0.03 | |||
| Any mutation | 0.0071 | 0.0078 | .364 | 1.65 | |||
| Any mutation >10% | 0.0015 | 0.0107 | .885 | 0.35 | |||
| Monocytes | Age + smoking | log10 | DNMT3A | 0.0061 | 0.0098 | .535 | 1.42 |
| DNMT3A >10% | 0.0068 | 0.0148 | .644 | 1.59 | |||
| DNMT3A VAF continuous | 0.0003 | 0.0006 | .630 | 0.07 | |||
| TET2 | 0.0067 | 0.0132 | .614 | 1.54 | |||
| TET2 >10% | −0.0038 | 0.0172 | .824 | −0.88 | |||
| TET2 VAF continuous | 0.0015 | 0.0006 | .013 | 0.35 | |||
| Any mutation | 0.0068 | 0.0082 | .405 | 1.58 | |||
| Any mutation >10% | 0.0038 | 0.0112 | .737 | 0.87 | |||
| Neutrophils | Age + smoking | log10 | DNMT3A | 0.0183 | 0.0096 | .057 | 4.30 |
| DNMT3A >10% | 0.0122 | 0.0144 | .397 | 2.84 | |||
| DNMT3A VAF continuous | 0.0006 | 0.0006 | .294 | 0.14 | |||
| TET2 | −0.0303 | 0.0127 | .017 | −6.73 | |||
| TET2 >10% | −0.0417 | 0.0166 | .012 | −9.15 | |||
| TET2 VAF continuous | −0.0011 | 0.0006 | .053 | −0.26 | |||
| Any mutation | 0.0007 | 0.0079 | .929 | 0.16 | |||
| Any mutation >10% | −0.0058 | 0.0108 | .595 | −1.32 | |||
| Platelets | Age | log10 | DNMT3A | 0.0014 | 0.0068 | .831 | 0.33 |
| DNMT3A >10% | −0.0022 | 0.0102 | .827 | −0.51 | |||
| DNMT3A VAF continuous | −0.0002 | 0.0004 | .702 | −0.03 | |||
| TET2 | −0.0124 | 0.0092 | .175 | −2.82 | |||
| TET2 >10% | −0.0256 | 0.0120 | .033 | −5.73 | |||
| TET2 VAF continuous | −0.0008 | 0.0005 | .073 | −0.19 | |||
| Any mutation | 0.0032 | 0.0056 | .572 | 0.73 | |||
| Any mutation >10% | −0.0017 | 0.0077 | .829 | −0.38 | |||
| MCV | Age + smoking | none | DNMT3A | −0.0710 | 0.2686 | .791 | −0.08 |
| DNMT3A >10% | −0.4800 | 0.4034 | .234 | −0.52 | |||
| DNMT3A VAF continuous | −0.0118 | 0.0157 | .455 | NA | |||
| TET2 | −0.3377 | 0.3535 | .340 | −0.37 | |||
| TET2 >10% | −0.7134 | 0.4616 | .123 | −0.78 | |||
| TET2 VAF continuous | −0.0162 | 0.0163 | .320 | NA | |||
| Any mutation | −0.0263 | 0.2192 | .905 | −0.03 | |||
| Any mutation > 10% | −0.3206 | 0.3011 | .287 | −0.35 | |||
| XCI ratio skewing | Age | none | DNMT3A | 0.0227 | 0.0114 | .046 | 9.56 |
| DNMT3A >10% | 0.0707 | 0.0169 | 3.10E-05 | 29.84 | |||
| DNMT3A VAF continuous | 0.0026 | 0.0006 | 5.67E-05 | NA | |||
| TET2 | 0.0697 | 0.0144 | 1.55E-06 | 29.38 | |||
| TET2 >10% | 0.0924 | 0.0187 | 9.55E-07 | 38.94 | |||
| TET2 VAF continuous | 0.0034 | 0.0007 | 4.20E-07 | NA | |||
| Any mutation | 0.0375 | 0.0092 | 4.76E-05 | 15.80 | |||
| Any mutation >10% | 0.0746 | 0.0124 | 2.53E-09 | 31.42 | |||
| Telomere length | Age | log10 | DNMT3A | −0.0104 | 0.0096 | .279 | −2.37 |
| DNMT3A >10% | −0.0155 | 0.0147 | .292 | −3.51 | |||
| DNMT3A VAF continuous | −0.0008 | 0.0006 | .143 | −0.19 | |||
| TET2 | −0.0254 | 0.0129 | .049 | −5.68 | |||
| TET2 >10% | −0.0073 | 0.0172 | .673 | −1.66 | |||
| TET2 VAF continuous | −0.0010 | 0.0007 | .129 | −0.23 | |||
| Any mutation | −0.0205 | 0.0079 | .010 | −4.60 | |||
| Any mutation >10% | −0.0196 | 0.0110 | .076 | −4.40 |
Correction for multiple testing (P < .0024).
 , standardized regression coefficient; MCV, mean corpuscular volume; NA, not applicable; SE, standard error; Trans., transformation.
, standardized regression coefficient; MCV, mean corpuscular volume; NA, not applicable; SE, standard error; Trans., transformation.
XCI skewing is an indirect measure of clonality and quantitative correlation with acquired somatic mutations is expected. XCI skewing in PMNs (controlled for age) was correlated with DNMT3A or TET2 and for all mutated individuals (Table 1). DNMT3A mutation correlated with XCI only for subjects with VAF >10% (+29.84%, P < .0001, Table 1) or when VAF was used as a continuous variable (P < .0001, Table 1). This supports an almost direct correlation between the number of DNMT3A mutated cells and change in XCI ratios. TET2 correlated with XCI skewing when all VAF were considered (+29.38%, P < .0001); this correlation was maintained for VAF >10% (+38.94, P < .0001). This indicates that TET2 mutation occurred in some individuals that already had a certain degree of XCI skewing.
Effect of DNMT3A and TET2 mutational status on blood counts and indices
The most important objective of this study was to determine the influence of acquired mutations on hematopoiesis. Because the vast majority of mutations occurred in DNMT3A or TET2, we had sufficient power to analyze their impact independently. We also performed analysis for all mutated individuals grouped together. Importantly, we performed qualitative and quantitative analyses. For the latter, we used an arbitrary VAF cutoff of >10% (>20% mutated cells) and also performed analysis using VAF as a continuous trait. All end points were controlled for covariates documented to influence the end point (see “Methods” for further details). DNMT3A mutation status did not affect any hematological end points. Even mutation at position R882 in DNMT3A had no impact on blood counts (supplemental Table 3). TET2 mutations were associated with a decrease in PMN counts. A trend was observed for all TET2 mutated individuals (−6.73%, P = .017, Table 1), whereas the neutropenic effect increased for subjects with VAF >10% (−9.15%, P = .012, Table 1). There was also a trend toward thrombocytopenia (−5.73, P = .033) only in individuals with a VAF >10%. Although none reached the .0024 significance level after Bonferroni correction, we consider these observations of interest. No effect was found on monocyte counts or MCV.
Even in subjects with multiple mutations (n = 50), we documented no impact on hematological end points. Of interest, subjects with both DNMT3A and TET2 mutations had no neutropenia (data not shown). Results from blood indices of individuals with multiple high-frequency mutations (≥2 mutations in any gene, at least 1 >10% VAF, n = 30; supplemental Table 4) confirms the absence of any pathological hematological phenotype with the exception of 2 individuals: 1 harboring both a TP53 and a TET2 mutation who had normochromic normocytic anemia, and 1 with double mutation in TET2 and NRAS who had mild anemia and thrombocytopenia.
Discussion
The recent demonstration10,11,14,20 that mutations in genes associated with hematological cancers occur in the normal aging population raised tremendous interest and concerns alike. This discovery represents a unique opportunity to decipher the multistep pathogenesis of myeloid cancers, potentially paving the way for novel diagnostic, preventive, and therapeutical strategies. However, current uncertainty about the risk of carrying these mutations led to the creation of a new diagnostic entity coined clonal hematopoiesis of indeterminate potential.21 The goal of this study was to gain more insight into the etiology and biological consequences of these mutations.
The whole genome approaches used by Xie et al,20 Genovese et al,10 and Jaiswal et al11 constituted the first appropriate step in identifying genes associated with clonal hematopoiesis. However, these methods lack analytical sensitivity and have limited gene coverage. We reasoned that greater precision is essential to document the exact prevalence of these mutations and the relative proportion between affected genes. Increased precision is also necessary to make valid correlations with different biologic end points. Using the described methodology, we identified a two- to threefold higher prevalence of mutations and a different proportion between affected genes than previously reported.10,11,20 Importantly, the majority of mutations (92.8%) involved DNMT3A or TET2, 2 epigenetic modifiers. This difference is explained mainly by better gene coverage and sensitivity. In the previously cited studies, only exon 3 of TET2 was efficiently captured.10,11 In this study, 57.5% (77/134) of the documented TET2 mutation resided outside exon 3. This may also be the case with DNMT3A, which was covered at 100%. Our methods achieved substantially higher sensitivity because of depth of sequencing and that we sequenced DNA of PMN from fractionated cells as opposed to whole blood. The limitation of a targeted gene approach is the number of candidate genes analyzed and their selection. Our panel missed a few genes that were recurrent in other studies, but at low frequencies. We specifically missed PPM1D, BCOR, GNB1, and GNAS. Based on previous studies,10,11,20 these genes would have collectively accounted for a prevalence of 0.44% or 11/358 subjects with mutation in our cohort. This would not have affected the conclusions of this study. Our approach did not allow to identify nondriver mutations, which have been recently shown to be several times more prevalent that driver mutations.32 However, the role of these mutations, which are identified with whole-genome sequencing, on hematopoiesis and cancer progression is uncertain at this time.
The increased sensitivity did not bias our study toward the identification of large numbers of subjects with small clones. In fact, the average size of the clone (VAF) was 14.3%, which corresponds to 28.6% of cells originating from mutated stem cells. Almost one-half of the mutated cohort (48.7%; 169/347) had a VAF ≥10%. Some subjects had more than 1 mutation (50/347); these individuals were older and had a higher VAF than those with a single mutation. The most frequent second mutation was 1 in the same gene (TET2-TET2 or DNMT3A-DNMT3A) or DNMT3A combined with TET2. In the latter situation, no specific order of acquisition was identified.
The documented mutations in DNMT3A and TET2 are similar to those documented in cancer, with the exception of the DNMT3A hot-spot mutation at position R882,33,34 which accounted for only 9.1% (21/230) of all DNMT3A mutations in contrast to its higher prevalence in MDS (23%) or AML (58%).35 For TET2, we were able to document a loss of function that reduced the 5hmC/5mC ratio proportionally to the VAF of the mutation, suggesting that all TET2 mutations have a similar functional impact.
As previously demonstrated, the principal factor associated with acquiring mutations is age.10,11,14,20 The global prevalence ranged from 6.3% in individuals 55 to 59 years of age to 42.9% in those ≥90 years of age. This age effect is more important for TET2 than for DNMT3A (odds ratio of acquiring a mutation 1.0937 vs 1.0404, respectively) and explains why TET2 is the most prevalent mutated gene after 85 years of age. We hypothesize that small (undetectable) TET2 clones are differentially favored with senescence. There may also be a survival advantage associated with TET2 compared with DNMT3A. Surprisingly, mutations in TET2 or DNMT3A were not associated with cardiovascular diseases or hypertension, contradicting previous studies and possibly explained by a female-based population (less affected by cardiovascular diseases). However, mutation in TET2 was associated with self-reported asthma-COPD similarly to the study of Zink et al.32
We then tried to estimate the relative proliferative advantage conferred by a mutation in DNMT3A vs TET2 on hematopoietic stem cells. In the absence of prospective resampling of the cohort, which would be required for definitive conclusions, we hypothesized that the relationship between clone size and specific age at sampling would be a surrogate of time-dependent clonal expansion. This assumption is supported by the similarities between different types of mutations across age groups. There is a yearly increase in the estimated mean VAF of 3.95% for DNMT3A and of 9.98% for TET2 (P < .0001). This may indicate a greater proliferative rate for TET2 mutation over DNMT3A, but more importantly, that these clones outperform the normal nonmutated stem cells. This is in contrast, however, with the work of Young et al showing that DNMT3A and TET2 mutations were present at low frequency in almost all younger individuals and that the clone size was stable over time.36 There are several hypotheses for this discordance, 1 being that the growth advantage is very small and that it takes several decades to reach significant clone expansion. However, this would not explain why only some individuals eventually progress to a detectable clone (VAF ≥2%). An alternate hypothesis is that the growth advantage is negligible compared with younger, more robust WT stem cells. As individuals age, the robustness of the nonmutated stem cell may decrease and the relative advantage conferred by the mutation may become more important. Because not all individuals develop a significant clone, this implies an interindividual variation in the competiveness of nonmutated stem cells with the mutated ones. In support of a relative small growth advantage, we did not document a reduction of age-adjusted TL in mutated individuals vs controls. We have previously documented that TL was severely reduced in subjects with JAK2 V617F positive MPN even at low VAF.31
We examined the impact of acquired mutations on blood counts and indices. Remarkably, there was no statistically significant impact on WBC, Hb, lymphocytes, monocytes, PMNs, platelets, or MCV. The only trends were documented in TET2 mutants with a reduction in PMNs (9%) and platelets (5%). Furthermore, even subjects with combined mutations did not have modified blood parameters. This indicates, without a doubt, that despite acquired mutations with aging, neither significant cytopenic nor proliferative effects are discernible and blood homeostasis is ultimately maintained. This observation is in contrast with murine models deficient for Tet2 or Dnmt3a.37-41
The fact that part of the cohort was recruited in sib-ships allowed us to investigate the heritability of mutations. We documented a significant familial risk of 2.7-fold for TET2 but not for DNMT3A. There was no germline mutation in TET2, and the specific TET2 mutations were different between siblings, suggesting genetic susceptibility loci. This is similar to what has been demonstrated in MPN, where familial aggregation has been identified and some predisposing candidate genes identified.42-44
The oncogenicity of these mutations is a matter of concern. This is supported by the ∼10-fold odds ratio of developing a hematological cancer reported by Jaiswal et al11 and Genovese et al10 and the consistent documentation of preleukemic mutations in DNMT3A or TET2 in leukemia or MDS patients.45-47 This risk seems to be influenced by the VAF and the number of mutation.32 However, it is important to note that of the combined 20 patients that developed hematological cancers in these studies, only 1 had a TET2 mutation (85 years of age; diffuse large cell lymphoma of the intestine), and 4 had DNMT3A mutations (only 1 with a confirmed myeloid cancer). Further, the recent documentation that DNMT3A and TET2 mutations are ubiquitous at low frequencies in middle-aged individuals indicates astronomically low oncogenic penetrance.36 Finally, a recent study demonstrated uncompromised 10-year survival in elderly subjects (>85 years of age) carrying such mutations.48 Taken together, these would not support a significant transformation risk for TET2 and to a lesser extent DNMT3A. This raises at least 2 questions. First, why do certain individuals effectively progress to cancer? The specific gene involved, the VAF, the number of different mutations, specific epigenetic marks, and stochastic events are all valid end points that need to be evaluated prospectively. Second, why do these mutations (specifically DNMT3A and TET2) arise and expand in the first place? We hypothesize that the mutated stem cells expand to compensate for failing senescent stem cells to provide normal hematological output.
In summary, we document that driver gene mutation–associated clonal hematopoiesis principally involves DNMT3A and TET2 genes. We did not document significant impact on blood counts except for a trend toward decreased neutrophils in subjects with TET2 mutation. TET2 mutations show familial aggregation suggesting a predisposing locus. Prospective evaluation of this cohort and sequential analysis of blood specimens will provide informative insights into the oncogenic penetrance of these mutations and potentially identify factors contributing to transformation. Finally, the role of these acquired mutations remains unclear and a matter of high interest.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Alain Tessier (Centre for Biological Applications of Mass Spectrometry, Concordia University) for mass spectrometry analysis.
This work was supported by a grant from the Canadian Institutes of Health Research and by the Leukemia Lymphoma Society of Canada.
Authorship
Contribution: M.B. contributed to project conception and coordination, performed and analyzed next-generation sequencing experiments with V.B. and G.L., generated all figures, tables, and supplementary material, and cowrote the paper; M.-P.D. supervised the statistical analyses; S.P. performed the statistical analyses with Y.F.Z. and A.B.; L.M. and N.S. interpreted data and edited the manuscript; and L.B. designed the project, obtained funding, interpreted data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Lambert Busque, Hôpital Maisonnneuve-Rosemont, Montreal, QC H1T 2M4, Canada; e-mail: lbusque.hmr@ssss.gouv.qc.ca.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal