

Abstract
Follicular lymphoma (FL) is a clinically and molecularly highly heterogeneous disease. Most patients achieve long-lasting remissions and have excellent overall survival (OS) with current treatment. However, ∼20% of patients have early progression of disease and short OS. At present, therapies are not guided by individual risk or disease biology. Reliable tools for patient stratification are urgently needed to avoid overtreatment of low-risk patients and to prioritize alternative approaches in high-risk patients. A rapidly expanding repertoire of promising therapeutic options is available for clinical evaluation; however, the numbers of patients with FL and the resources to conduct adequately powered trials are limited. Recent studies have shown that gene mutations can serve as prognostic and/or predictive biomarkers, in particular when integrated into composite risk models. Before translating these findings into routine clinical practice, however, several challenges loom. We review aspects of “clinicogenetic” risk model development and validation that apply to FL and more generally to other cancers. Finally, we propose a crowdsourcing effort that could expedite the development, validation, refinement, and selection of risk models. A new era of collaboration and harmonization is required if we hope to transition from empiric selection of therapeutics to risk-based, biology-guided treatment of patients with FL.
Introduction
Follicular lymphoma (FL) is among the most common non-Hodgkin lymphomas worldwide, accounting for approximately one-quarter of new cases in the United States and Europe.1,2 It may well be the most prevalent of all non-Hodgkin lymphomas, due to its long natural history and low cure rates. FL is a prototypical indolent lymphoma, and median overall survival (OS) now exceeds 18 years with modern treatments.3 At the same time, FL is a highly heterogeneous disease, and a subset of patients has remarkably poor outcome.4,5
Less than one-quarter of patients present with nonbulky, limited-stage disease. Both the National Comprehensive Cancer Network6 and European Society for Medical Oncology7 recommend radiotherapy in these patients with curative intent. However, only one-half who undergo radiotherapy achieve long-term remissions, and comparable outcomes have been reported with observation or single-agent rituximab.8,9
The remaining patients are diagnosed with advanced-stage or bulky, limited-stage disease and are still considered incurable with conventional therapies.10,11 Patients with asymptomatic disease may not need treatment for several years. We cannot reliably predict whether or when individual patients will progress to the point where treatment is required. Even among patients who require immediate treatment due to symptomatic, rapidly progressive, and/or large burden disease, clinical course remains highly variable. Currently, these patients uniformly receive immunochemotherapy.12 The specific choice of treatment regimen, administration of maintenance and/or consolidation, and even disease monitoring are largely driven by center/physician preference rather than by individual risk or disease biology. Although lamentable, this is not unreasonable, as no approach has clearly demonstrated the ability to prospectively and reliably identify patients who will experience either early progression of disease, which we define here as within 24 months after immunochemotherapy (POD24)4,5 or histologic transformation to aggressive lymphoma.13
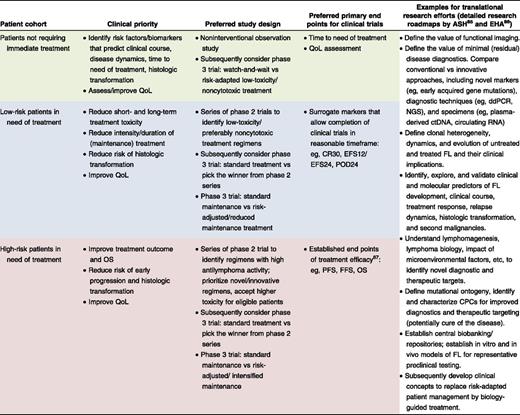
Clearly, reliable tools for patient stratification are urgently needed to both avoid overtreatment of low-risk patients and to select high-risk patients for novel strategies. A formidable repertoire of promising therapeutic options is available for clinical evaluation in patients with FL.14 Resources to conduct adequately powered studies with sufficient follow-up are limiting, as are the numbers of patients with FL willing to participate in clinical trials. Stratification of patients by individual risk and disease biology will be crucial to adequately address distinct clinical priorities, define suitable study end points, interrogate individual treatment strategies, and conduct strata-tailored translational research (Table 1). Here, we will discuss the promises and challenges of integrating gene mutations into clinically applicable stratification algorithms for patients with FL. The same concepts are largely applicable to other assays capturing, for example, genomic, epigenomic, transcriptomic, proteomic, metabolomic, or microenvironmental alterations, as well as diseases other than FL.
Proposed strategies for clinical trials in patients with untreated advanced-stage FL
 |
|
ASH, American Society of Hematology; CPC, common progenitor clone; ctDNA, circulating tumor DNA; CR30, complete response rate at 30 mo; ddPCR, digital droplet PCR; EFS12/EFS24, event-free survival at 12/24 mo; EHA, European Haematology Association; FFS, failure-free survival; FL, follicular lymphoma; NGS, next-generation sequencing; OS, overall survival; PFS, progression-free survival; POD24, progression of disease within 24 mo; QoL, quality of life.
Gene mutations as prognostic/predictive biomarkers
FLs frequently harbor mutations not only in genes that encode for epigenetic modifiers (including KMT2D, CREBBP, EP300, and EZH2), but also in genes encoding for transcription factors, kinases, and other signaling molecules (supplemental Table 1, available on the Blood Web site).15-27 To some extent, FL is a genetically homogeneous disease: ∼90% of cases harbor translocations involving BCL2, 80% have loss-of-function mutations of KMT2D, and 80% have mutations of CREBBP or its homolog EP300. On the other hand, hundreds of additional recurrent gene mutations have been reported in FL, which are likely to have distinct implications on the biology28-40 and the clinical course41-49 of the disease (supplemental Table 1).
In the following sections, we discuss the use of gene mutations as biomarkers, which can be either prognostic, or predictive, or both. Conceptually, prognostic biomarkers predict patient outcomes independent of treatment, whereas predictive biomarkers predict the effects from a specific treatment in biomarker-positive patients compared with biomarker-negative patients.50 Making the distinction between prognostic and predictive requires formal statistical testing (interaction analysis) of at least 2 comparison groups, however, such testing for gene mutations in FL is pending.
Single-gene mutations
Mutations in a few individual genes have been associated with treatment outcome among patients with FL. Most notably, TP53 mutation predicted inferior outcome both in the prerituximab51 and rituximab eras.41 In unselected patient cohorts, TP53 mutations are present in <5% of patients at initial diagnosis. Consistent with their adverse effect on prognosis, TP53 mutations were found to be enriched in patients with early progression of disease5,52 or histologic transformation.22,47,48,52
The histone methyltransferase EZH2, which functions as the catalytic subunit of the polycomb-repressive complex 2 (PRC2), has been identified as both a biomarker and therapeutic target. Mutations in EZH2 (primarily affecting residue Y641) are present in approximately one-quarter of all FLs at initial diagnosis.19,53 Heterozygous EZH2 mutations are gain of function by increasing trimethylation of histone H3 lysine 27 (H3K27me3),33 an epigenetic mark of repressed gene expression, which promotes lymphomagenesis in vivo in combination with BCL2 overexpression.35 EZH2 mutations have been consistently associated with favorable treatment outcome in studies of homogenously treated patients who received frontline immunochemotherapy for advanced, symptomatic FL. Analyses from both the PRIMA trial and the Lunenburg Consortium demonstrated that EZH2 mutations were associated with improved outcome.43,45 Interestingly, the PRIMA study also suggested that cases with EZH2 copy number gains, although less frequent, harbor transcriptional profiles and have treatment outcomes highly similar to EZH2-mutant cases.43
The data for other single-gene mutations as biomarkers are more controversial (supplemental Table 1). For example, mutations affecting TNFRSF14 at initial diagnosis were associated with shorter survival in nonuniformly treated patients in one study46 but not in others.45,49 Also, coding sequence mutations in BCL2 were associated with shorter survival in one study42 but not in others.41,44 BCL2 mutations were also suggested as a surrogate marker for activation-induced cytidine deaminase (AID)–mediated genetic instability, which may predict an increased risk of histologic transformation.42
Integrating multiple-gene mutations into composite risk models
In contrast to individual gene mutations, combining multiple-gene mutations into risk models could capture the complex and interdependent interactions between distinct gene mutations, which are still largely unknown. Multivariate modeling allows for the integration of gene mutations, even if univariate testing did not indicate a statistically robust effect on treatment outcome (and vice versa). Furthermore, composite risk models can integrate additional variables; these could include clinical factors (like those in the Follicular Lymphoma International Prognostic Index [FLIPI] or performance status), germ line polymorphisms, disease-related alterations (eg, in DNA copy number, the epigenome, transcriptome, proteome, or metabolome), microenvironment-related factors (eg, cytokine milieu, abundance of tumor cells, or infiltrating immune cell subsets), or as yet unidentified factors.
In 2015, we published a clinicogenetic risk classifier called the m7-FLIPI, which integrates the mutation status of 7 genes, the FLIPI, and Eastern Cooperative Oncology Group (ECOG) performance status. We began by genotyping samples for mutations in 74 genes recurrently mutated in FL and then generated a multivariate model irrespective of the results from univariate testing. Applying the m7-FLIPI to individual patients involves genotyping of the 7 genes for nonsilent mutations that occur at a variant allele frequency (VAF) of 10% or more. The cumulative risk score is calculated by adding individual predictive weights that either increase (mutations in EP300, FOXO1, CREBBP, and CARD11) or decrease (mutations in EZH2, ARID1A, and MEF2B) the cumulative risk score. A predefined threshold for the cumulative risk score distinguishes high-risk and low-risk patients (online tool at http://www.glsg.de/m7-flipi/).
The m7-FLIPI improved risk stratification for failure-free survival (FFS) and OS41 and was predictive for POD245 in both a discovery cohort of clinical trial patients and a validation cohort of nontrial patients who uniformly received standard regimens of immunochemotherapy as frontline treatment of symptomatic FL. Specifically, the m7-FLIPI identified 22% (discovery cohort) and 28% (validation cohort) of patients as high-risk with 5-year FFS rates of 38% and 25%, and 5-year OS rates of 65% and 42%, respectively.41 The m7-FLIPI outperformed the FLIPI alone in both cohorts and had a high specificity for POD24 (the true negative rate was 79% and 86%, respectively).
The major shortcoming of the m7-FLIPI was that it only classified about one-half of patients with POD24 as high-risk. To specifically address this, we generated a subsequent model, the POD24-PI (POD24–Prognostic Index), which consisted of only 3 gene mutations (EP300, FOXO1, and EZH2) along with the FLIPI. Although the POD24-PI had higher sensitivity for predicting POD24, it came at the expense of lower specificity.5 Thus, the combination of gene mutations and clinical factors improves prognostic accuracy beyond clinical factors alone but, as one would expect, is not perfect.
Validation and exploration in addition cohorts
Validation of a prognostic algorithm within a single validation cohort does not ensure that the algorithm is robust across a range of different patient populations. However, it is important to clarify whether those additional patient populations comply with the same definitions and clinical end points used to establish the algorithm. For example, the m7-FLIPI was developed and validated only in patients with FL grade 1, 2, or 3A, advanced-stage or bulky disease considered ineligible for curative irradiation, and symptomatic disease requiring systemic treatment. All patients received a combination of rituximab and chemotherapy (either cyclophosphamide, doxorubicin, vincristine, and prednisone [CHOP] or cyclophosphamide, vincristine, and prednisone [CVP]) as frontline treatment, and were only analyzed if biopsies were obtained within 1 year of beginning treatment. The latter requirement was intended to minimize the confounding effects from any genetic drift that might occur in untreated FL between the time of biopsy and the time of treatment (eg, by aberrant somatic hypermutation54 ). The primary end point was FFS, which is calculated from time of treatment initiation and differs from progression-free survival (PFS) by also including insufficient response (ie, less than a partial response) as an event; that was felt to be most appropriate for patients with symptomatic disease in need of treatment. We used exon-capture sequencing, paraffin-embedded tissue, and a minimum VAF of 10% to call an individual mutation.
If the patient population and/or approach being tested in a subsequent assessment of the m7-FLIPI differs from these strict definitions, then the question being addressed is not whether the m7-FLIPI “passes” or “fails” validation. This distinction is not meant to keep investigators from exploring the m7-FLIPI on different types of cohorts or using different approaches, which are important steps of assessing its extendibility.
It goes without writing that validation studies and exploratory analyses must also be adequately powered. Figure 1 shows a nomogram visualizing the relationship between the hazard ratio (HR) of a given risk model, the fraction of patients classified to be high-risk, and the numbers of events required for adequate power. The requirement for an adequate number of events (vs an adequate number of patients) has a number of practical implications. A cohort may not be sufficiently powered upon initial assessment but, with the accumulation of events, becomes adequately powered (ie, there is no need to increase the cohort size itself). Also, the known or expected HR of risk models may vary with factors like the efficacy of the treatment regimen: the higher the HR, the lower the number of events needed for validation studies, and vice versa. And finally, the lowest number of events is needed for cohorts with a 1:1 ratio of patients classified to be low- or high-risk (Figure 1).

Nomogram for known or expected HRs of a risk model, fractions of patients identified as high-risk, and numbers of events needed for a power of 0.9. The practical implications of these relationships can be exemplified as follows: validation of the m7-FLIPI in a R-CVP–treated cohort (HR of ∼3.6) requires fewer FFS events compared with a R-CHOP–treated cohort (HR of ∼2).41 Similarly, a smaller number of events will be required if the fraction of high-risk patients is approaching 0.5, for example, as expected when analyzing only patients with high-risk FLIPI.5
Nomogram for known or expected HRs of a risk model, fractions of patients identified as high-risk, and numbers of events needed for a power of 0.9. The practical implications of these relationships can be exemplified as follows: validation of the m7-FLIPI in a R-CVP–treated cohort (HR of ∼3.6) requires fewer FFS events compared with a R-CHOP–treated cohort (HR of ∼2).41 Similarly, a smaller number of events will be required if the fraction of high-risk patients is approaching 0.5, for example, as expected when analyzing only patients with high-risk FLIPI.5
Krysiak et al recently published a study that significantly expanded the catalog of recurrently mutated genes in FL.17 This study also explored the m7-FLIPI in a clinically heterogeneous cohort of 81 patients, of whom 58 received treatment within 1 year of diagnosis. The m7-FLIPI reclassified almost one-half of patients with high-risk FLIPI as low-risk by m7-FLIPI (7 of 16), and had a HR for PFS of 1.9. The differences in patient population between this cohort and those within the original m7-FLIPI publication41 indicate that the m7-FLIPI may retain prognostic ability across less rigidly defined populations of FL. However, this study was not sufficiently powered to reach statistical significance, so one could inappropriately conclude that the m7-FLIPI “failed” to demonstrate prognostic utility in this cohort.
At the 14th International Conference on Malignant Lymphoma in Lugano (14-ICML), Huet et al reported on the performance of clinicogenetic risk models in patients from the PRIMA trial.55 In patients who received immunochemotherapy followed by rituximab maintenance, the m7-FLIPI had a HR of 2.9 for PFS and outperformed the FLIPI (HR, 2.0), but not the FLIPI-2 (HR, 3.7).55,56
Standardization and harmonization
Discrepancies across studies and failure to reproduce or validate earlier findings may also be due to different technical and analytical approaches. No consensus has been established as to the optimal DNA isolation, sequencing, and analysis approaches. DNA from formalin-fixed paraffin-embedded biopsy specimens is fragmented and chemically modified, and can be challenging to sequence. Often, matched germ line samples are not available to definitively distinguish somatic from germ line variants. The tumor cell fractions in biopsies may be low, which reduces the identification of subclonal variants. Evolving data further indicate that a single biopsy is unlikely to fully represent the spatial heterogeneity and clonal hierarchies of the disease,57 which can further affect the likelihood of capturing prognostically relevant mutations.
Recent data suggest that genotyping of circulating tumor DNA (ctDNA) could better capture mutational heterogeneity.58 Extensive studies of sequencing ctDNA are needed (and several are under way) to determine whether this approach will supplant more traditional biopsies, which would also greatly increase the opportunity to serially sample patients before, during, and after treatment. Minimally invasive serial sampling of ctDNA has tremendous clinical potential, including the evaluation of genetic drift over time in untreated FL, treatment responses and clonal dynamics, detection of residual or emerging treatment-resistant subclones, and even detecting and predicting histologic transformation.58-61
Harmonization across institutions is essential for generating highly reproducible and reliable approaches that are applicable to clinical decision-making. Funding and other material support is needed to help compare, optimize, and establish standards for patient specimens, sequencing methods, and bioinformatic pipelines.
Linking mutation biology to risk
The m7-FLIPI was intentionally designed as a straightforward clinical tool, and therefore gene mutations were integrated solely as binary variables (ie, presence or absence of a nonsilent mutation). Classifying genes in this binary fashion is an oversimplification that does not account for the multidimensionality of available sequencing data. The location and type of individual mutations, their presence as clonal or subclonal mutations, and other factors may affect their weighting within clinicogenetic risk models.
Implications of mutation localization and type. The m7-FLIPI is clearly biased toward genes harboring either mutational hotspots, such as EZH2 (97% of mutations at Y641 or A677), FOXO1 (90% in exon 1), MEF2B (100% within the N-terminal domain), or CARD11 (80% affecting the coiled-coil domain), or a distinct mutation type, such as primarily disruptive alterations in ARID1A.41 In contrast, CREBBP harbors at least 2 different types of mutations: the more common missense mutations clustered within the histone acetyltransferase domain, and the less frequent truncating mutations spread out across the whole coding region (supplemental Figure 1). CREBBP acts as an epigenetic modifier by acetylating not only lysine residues on histone 3 (H3), but also nonhistone proteins such as TP53, BCL6, and FOXO1.16,62 Evolving data suggest that specific CREBBP mutations are associated with distinct biology.20,32 It is possible that the different types of CREBBP mutations are also associated with distinct clinical outcomes, and that its comparatively low predictive weight in the m7-FLIPI results from averaging these effects.
Implications of zygosity. Genes can harbor mutations in 1 or both alleles, and also be affected by structural alterations (deletions, amplifications, or rearrangements). The resulting (mutant) gene-dosage effects can be functionally and clinically relevant. For example, the tumor suppressor gene TNFRSF14 is frequently affected by loss-of-function mutations (∼20%-30% of cases) and/or deletions of its chromosomal region at 1p36 (∼20% of cases).40 In 1 study, TNFRSF14 alterations were associated with inferior outcome, and shortest survival was seen with combined deletion and mutation of the remaining allele.46
Implications of clonality, molecular hierarchy, and mutational ontogeny. In chronic lymphocytic leukemia, the presence of subclonal driver mutations such as SF3B1 or TP53 has been shown to be an independent risk factor for rapid disease progression.63 This suggests that low VAF mutations may be prognostically relevant in FL as well, but a full analysis has not yet been published. As noted in “Integrating multiple-gene mutations into composite risk models,” mutations with VAF <10% are not scored in the m7-FLIPI.
At the other end of the spectrum, some highly recurrent gene mutations such as CREBBP and EZH2 are mostly clonal.19,20 Several publications have now established the concept of common progenitor clones (CPCs), by identifying a set of shared gene mutations in pairs of FL at initial diagnosis and at relapse and/or transformed FL.20-22,52 As expected, these (inferred) CPCs were enriched particularly for mutations in epigenetic regulators. Conceptually, as CPCs give rise to relapsed and/or transformed FL (mostly by divergent evolution), CPC-defining mutations represent highly promising candidates for minimal residual disease diagnostics (eg, using ctDNA) and as therapeutic targets.
Evolving evidence from transgenic models and patients suggests that the sequence of mutation acquisition at different stages of hematopoietic development has biological and clinical implications. In mice, genetic deletion of Crebbp and Kmt2d in early, antigen-naive B cells led to a more pronounced phenotype compared with inactivation at later stages, that is, in more mature germinal-center B cells.28,29 We have previously shown that FL-associated mutations can be acquired many years before patients develop clinically apparent manifestations,64 with a subset of mutations detectable in hematopoietic progenitor cells.65 In Langerhans cell histiocytosis, for example, clinically aggressive/high-risk disease arises from acquisition of the BRAF V600E mutation in hematopoietic precursor cells, whereas indolent/low-risk disease is driven by the identical gain-of-function gene mutation acquired in more mature, tissue-restricted precursor cells.66
Gene mutations are only one component of complex biological networks
Current targeted sequencing approaches undoubtedly do not comprehensively capture all biologically and clinically relevant gene mutations,67 but there is likely to be a point of diminishing return that has either already been reached or is rapidly approaching for common lymphomas like FL. As new technologies are increasingly applied in an unbiased manner to FL samples (eg, chromatin immunoprecipitation sequencing, transcriptome, proteome, metabolome, or single-cell analyses, and functional assays such as BH3 profiling68 ), these orthogonal approaches could be added to traditional DNA sequencing to improve prognostic models. Evolving evidence also suggests that biologically relevant mutations are not restricted to the coding genome: for example, mutations in regulatory elements have been shown to disrupt transcription factor binding and to attenuate the expression of their target genes.26 Mutant gene products have also been shown to serve as neoantigens in FL, capable of eliciting specific T-cell responses.69
Finally, the clinical impact of gene mutations may also depend on interactions with numerous other molecular alterations within the malignant cells and with components of the microenvironment.70 For example, recurrent mutations in the DNA-binding site of STAT6 have been shown to amplify the interleukin 4 (IL-4)–induced JAK-STAT signaling cascade.39,71 Thus, the biological and clinical impact of STAT6 mutations is expected to heavily depend on the abundance of follicular helper T cells, the major source of IL-4 in the microenvironment of FL,72,73 and thus may differ based on site of the disease, activity of a particular treatment regimen against follicular helper T cells, or even polymorphisms that affect IL-4 production and/or signaling.
Gene-expression profiling has the potential to capture the net “downstream” phenotype that results from these complex interactions, both within the malignant cells and with its microenvironment. Furthermore, proof-of-concept studies have demonstrated that immune cell–derived gene-expression signatures can predict treatment outcome,74,75 but due to technical challenges and methodological concerns76 this concept has not yet been translated into the clinics. Improved assays, such as the nCounter technology (NanoString) now allow robust and reliable gene-expression profiling from challenging formalin-fixed paraffin-embedded biopsy specimens, and are increasingly applied to various cohorts of patients with FL.77 It will be interesting to see if integration of prognostic gene-expression signatures into multivariable risk models will add to or even substitute for gene mutations for improved patient stratification in FL.
From risk-adapted to biology-guided treatment strategies
Unlike conventional cytotoxic chemotherapies, the response to molecular targeted therapies may be particularly predictable based on single-gene mutations. For example, mutant EZH2 confers a gain of function and thus may be a promising target for inhibitors of this enzyme.34 An interim analysis of an ongoing phase 2 trial of the EZH2 inhibitor tazemetostat in patients with relapsed lymphoma was presented at the 14-ICML: in 67 evaluable patients with FL, the overall response rate was 92% for patients with EZH2-mutant tumors (12 of 13), as compared with 26% for patients without detectable EZH2 mutations (14 of 54).78 Thus, EZH2 mutations were highly predictive of response but >50% of responders did not have EZH2 mutations. The latter may be related to EZH2 amplification43 or other alterations like ARID1A mutation, which can synthetically confer lethality with EZH2 inhibition.79,80 However, as mentioned earlier, EZH2 mutations are also predictive of favorable outcome after immunochemotherapy.41,43,45 This example illustrates that it remains to be determined whether molecular targeting strategies represent an advance on what could be seen with standard therapy, and whether their role will be to add to rather than replace conventional treatment approaches.
The obverse example is BTK inhibition, which is highly active against some B-cell lymphomas but only modestly active in FL.81 Interestingly, a number of gene mutations recurrently found in FL may confer ibrutinib resistance, such as NF-κB–activating mutations like CARD11 or TNFAIP3, or mutations in CXCR4 that activate AKT and ERK signaling.82,83 Thus, predictive algorithms that apply to regimens containing targeted therapies may require individualization; whether these can be established by modifying the existing algorithms like the m7-FLIPI or will require starting “from scratch” is unclear.
A vision for prognostic and predictive model development
The biggest challenge for any biomarker is to demonstrate clinical utility. We face a status quo where few if any biomarkers are used to drive treatment selection for patients with FL, despite the fact that massive data sets have been and continue to be accumulated from clinical trial patients and registries. To some extent, this is simply because novel agents are brought into trials at a pace that exceeds the ability to validate prognostic or predictive models within prospective clinical trials. As an example, by the time that data on rituximab plus CHOP or CVP (R-CHOP and R-CVP) regimens were mature enough for us to formulate the m7-FLIPI, the standard treatment of patients with advanced FL had switched to R-bendamustine at many centers.
How can we speed the pace of biomarker development and validation so that patient selection can move in parallel with therapeutic development? There may not be a definitive answer, but crowdsourcing could be helpful. In this regard, the American Society of Hematology (ASH) is establishing a Department of Registries with the goal of tackling problems that require information technology–based solutions. Patient, treatment, outcome, pathology, and genomic data will all be needed to make analyses most relevant to clinical application. Additional efforts and funding are needed to systematically implement comprehensive biobanking in clinical registries and trials. It will be essential to harmonize and standardize methodological approaches, as different DNA isolation protocols, different patient materials (eg, lymph node biopsy, bone marrow aspirate, or cell-free DNA), and different sequencing platforms may give disparate results. At the same time, investigators should be able to upload raw data (eg, FASTQ files) for analysis by a “standard bioinformatic pipeline” that is codeveloped by leaders in the field, iteratively improved under the auspices of an organization like ASH, and made available open source. This would help overcome much of the variability in analysis that currently affects sequencing efforts while extending more sophisticated bioinformatic analysis to investigators who currently lack access to it. One could envision computer scripts written in open-source language that are freely available from ASH. These scripts would be applied directly to clinical trial data (extant and future) in real time to either establish the most promising (clinicogenetic) risk model from a particular data set, or to assay existing algorithms against the data set and determine the most robust assay. All of these analyses would be centrally housed at ASH or with a similar honest broker so that researchers could immediately understand the performance of an individual risk model across diverse populations of either clinical trial–based or registry-based patients.
The major questions that remain in FL treatment (Table 1) are likely to be answerable in the next 5 years with the right approach. Much of the needed data already exists: an important step, as outlined by others in the larger context of cancer genomic data,84,85 is to make both the analysis of the data and the analysis of the analysis open to everyone.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Vindi Jurinovic and Donna Neuberg for statistical consulting.
O.W. was supported by the Max-Eder Program of German Cancer Aid (#110659), and the German Research Foundation (DFG SFB/CRC 1243, TP A11). D.M.W. was supported by a Leukemia & Lymphoma Society Scholar Award.
Authorship
Contribution: O.W. and D.M.W. wrote the manuscript.
Conflict-of-interest disclosure: O.W. receives consulting support from Roche, Incyte, and Cellestia, and research support from Roche and Novartis. D.M.W. receives consulting support from Novartis and Monsanto, and research support from Novartis, Roche, Surface Oncology, Abbvie, AstraZeneca, and Aileron.
Correspondence: Oliver Weigert, Department of Medicine III, Laboratory for Experimental Leukemia and Lymphoma Research (ELLF), University Hospital, LMU Munich, Max-Lebsche Platz 30, 81377 Munich, Germany; e-mail: oliver.weigert@med.uni-muenchen.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal