

Key Points
Our generated PDX models reflect the immunophenotypic, transcriptional, genetic, and functional heterogeneity of primary DLBCL.
The experimental and analytical approach will inform the development of additional PDX models and facilitate preclinical drug discovery.
Abstract
Diffuse large B-cell lymphoma (DLBCL) is a heterogeneous disease defined by transcriptional classifications, specific signaling and survival pathways, and multiple low-frequency genetic alterations. Preclinical model systems that capture the genetic and functional heterogeneity of DLBCL are urgently needed. Here, we generated and characterized a panel of large B-cell lymphoma (LBCL) patient-derived xenograft (PDX) models, including 8 that reflect the immunophenotypic, transcriptional, genetic, and functional heterogeneity of primary DLBCL and 1 that is a plasmablastic lymphoma. All LBCL PDX models were subjected to whole-transcriptome sequencing to classify cell of origin and consensus clustering classification (CCC) subtypes. Mutations and chromosomal rearrangements were evaluated by whole-exome sequencing with an extended bait set. Six of the 8 DLBCL models were activated B-cell (ABC)-type tumors that exhibited ABC-associated mutations such as MYD88, CD79B, CARD11, and PIM1. The remaining 2 DLBCL models were germinal B-cell type, with characteristic alterations of GNA13, CREBBP, and EZH2, and chromosomal translocations involving IgH and either BCL2 or MYC. Only 25% of the DLBCL PDX models harbored inactivating TP53 mutations, whereas 75% exhibited copy number alterations of TP53 or its upstream modifier, CDKN2A, consistent with the reported incidence and type of p53 pathway alterations in primary DLBCL. By CCC criteria, 6 of 8 DLBCL PDX models were B-cell receptor (BCR)-type tumors that exhibited selective surface immunoglobulin expression and sensitivity to entospletinib, a recently developed spleen tyrosine kinase inhibitor. In summary, we have established and characterized faithful PDX models of DLBCL and demonstrated their usefulness in functional analyses of proximal BCR pathway inhibition.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common non-Hodgkin lymphoma in adults, accounting for ∼40% of B-cell malignancies. DLBCL is a clinically and genetically heterogeneous disease with recognized subtypes based on morphology, transcriptional profiles, and multiple low-frequency genetic alterations including chromosomal translocations, somatic mutations, and copy number alterations (CNAs). In addition to typical DLBCLs, recognized morphologic and immunophenotypic subtypes include T-cell/histiocyte-rich large B-cell lymphoma and plasmablastic lymphoma (PBL), among others.1 The transcriptional heterogeneity of DLBCL is addressed by 2 classification schemes, cell of origin (COO) and consensus clustering classification (CCC).2-5 Both of these systems highlight specific aspects of DLBCL biology, suggest cancer cell dependencies, and identify rational therapeutic targets.
The COO classification defines DLBCLs that share certain features with normal B-cell subtypes and includes germinal center B-cell (GCB) and activated B-cell (ABC) types.6,7 GCB-type DLBCLs are tumors derived from light-zone germinal center B cells. MYC and BCL2 translocations are detected in 10% and 40% of these lymphomas, respectively.6 Additional genetic alterations that occur more frequently in GCB-type DLBCLs include gain-of-function mutations in the chromatin-modifying enzyme EZH28 and inactivating mutations of the Gα13 migration pathway components such as GNA13.9 In contrast, ABC-type DLBCLs are derived from a germinal center B cell committed to plasmablastic differentiation.6 ABC-type DLBCLs have increased nuclear factor-κB (NF-κB) activity and more-frequent genetic alterations of certain NF-κB pathway components, including MYD88 and CARD11.10-12 ABC-type DLBCLs also have more frequent somatic mutations of the genes encoding the immunoglobulin α or β subunits (CD79A or B) and associated alterations of B-cell receptor (BCR) surface expression and signaling.10 These tumors also exhibit alterations associated with a terminal differentiation block, such as PRDM1 (BLIMP1) deletion.13
Additional aspects of DLBCL functional heterogeneity are captured by the CCC system, which categorizes DLBCLs purely on the basis of the tumor transcriptome and defines BCR, oxidative phosphorylation (OxPhos), and host response (HR) subtypes.2,4,5 The BCR-type DLBCLs have more abundant expression of proximal components of the BCR pathway and increased reliance on proximal BCR signaling and survival pathways.5,14 These tumors include BCR-dependent ABC-type DLBCLs with high baseline NF-κB activity and BCR-dependent GCB-type DLBCLs with low baseline NF-κB activity that largely rely on spleen tyrosine kinase (SYK)/ phosphatidylinositol 3-kinase (PI3K)/protein kinase B signaling (AKT).5 The consequences of proximal BCR/SYK/PI3K pathway inhibition differ in BCR-dependent DLBCLs with high or low baseline NF-κB activity (ABC or GCB types, respectively). In BCR-dependent ABC-type DLBCLs, proximal BCR pathway inhibition decreases the abundance of NF-κB target genes, including antiapoptotic BCL2 family members such as BCL2-related protein A1 (BCL2A1, also known as BFL1/A1).5 In contrast, proximal BCR pathway inhibition induces the proapoptotic BH3 family member HRK in BCR-dependent GCB-type DLBCLs.5 BCR-type DLBCLs exhibit additional genetic alterations of proximal BCR pathway components, including copy gain of SYK and copy loss of the PI3K negative regulator PTEN.5 BCR-independent OxPhos–type DLBCLs exhibit enhanced mitochondrial energy transduction and selective reliance on fatty acid oxidation.4 HR–type DLBCLs have a characteristic inflammatory/immune cell infiltrate and include the morphologically defined subset of T-cell/histiocyte-rich B-cell lymphomas.2
Additional genetic features of DLBCL are not associated with transcriptionally defined subtypes, including frequent CNAs of p53/cell cycle pathway components and less common somatic mutations of TP53, gain-of-function mutations in the BCL6-modulator MEF2B, and perturbations of antigen presentation machinery and chromatin modification.6,15-17
Despite advances in the molecular and functional characterization of DLBCL, newly diagnosed patients are still largely treated with the same empiric rituximab plus cyclophosphamide/doxorubicin/vincristine/prednisolone (R-CHOP) regimen. Although up to 60% of DLBCL patients are successfully treated with this regimen, the remainder have limited therapeutic options and often succumb to their disease.18 Preclinical model systems that capture the genetic and functional heterogeneity of primary DLBCL are urgently needed. DLBCL cell lines and associated xenografts are routinely used to analyze rational targets and candidate inhibitors; however, these are imperfect models of the disease. For example, almost all DLBCL cell lines have inactivating somatic mutations of TP53, whereas only ∼20% of primary DLBCLs have this alteration.15,19-22
Although genetically engineered mouse models have revealed mechanistic insights into single genes and pathways,13,23-26 they do not capture the genetic, transcriptional, and functional heterogeneity of primary human DLBCL. Moreover, these mouse models have variable penetration and time of onset, making them suboptimal systems for drug development. Patient-derived xenograft (PDX) models27-32 of primary DLBCLs should overcome these limitations and provide more reflective and reliable models of the disease. However, establishing PDX models of DLBCL has been historically difficult. We have reevaluated and optimized the generation of large B-cell lymphoma (LBCL) PDX models using more-immunocompromised hosts and newer inoculation strategies. Herein, we report both the establishment and the genetic and biological characterization of a series of 9 human LBCL PDX models that capture the heterogeneity of the disease.
Patients, materials, and methods
Patient specimens and animals
DLBCL patient specimens were obtained from the British Columbia Cancer Agency with written informed patient consent, following a protocol approved by the Clinical Research Ethics Board of the University of British Columbia. Six- to 8-week-old male NOD SCID IL2Rγnull (NSG) mice (The Jackson Laboratory) were used for the establishment of PDX models. All related experimental protocols were approved by the University of British Columbia Animal Care Committee and the Dana-Farber Cancer Institute Animal Care and Use Committee.
Development of PDX models
Subrenal capsule PDX models were generated as previously described.33,34 Surgically removed patient tumor tissue was collected and stored in 4°C Hanks balanced salt solution supplemented with antibiotics and transported to the animal facility within 2 hours of surgery. Each fresh tumor sample was cut into multiple 1 × 2 × 3 mm3 pieces and kept in sterile Hanks balanced salt solution until it was xenotransplanted. Mouse surgery was performed in a biosafety level 2 laminar flow hood under sterile conditions. An incision of ∼1.0 cm was made along the midline of the back of an anesthetized mouse, followed by the opening of the mouse body wall above 1 kidney. Subsequently, the kidney was mechanically pressed through the incision. With the exteriorized kidney resting on the body wall opening, a 2- to 3-mm kidney membrane incision was made with a pair of spring-loaded scissors. Thereafter, a small pocket was created between the kidney membrane and parenchyma using a glass pipette dissector. Then, the primary tumor piece was inserted under the renal capsule and the kidney was gently eased back into the body cavity. The edges of the body wall and skin were aligned and closed with 5-0 surgical threads. Six to 8 male NSG mice were implanted with each primary LBCL.
After xenotransplantation, animals were evaluated regularly for palpable tumors. When tumors were palpable, the animals were euthanized, and tumors were excised and passaged to another cohort of animals. In the described PDX models, the median time to palpable tumor was 45 days (range, 18-257 days). Each graft was serially passaged 5 times by subrenal capsule transplant to generate a stable PDX model. For each LBCL PDX model, tumor tissue was excised, and DNA and RNA were extracted as previously described.15
Whole-transcriptome sequencing (RNA-seq)
Library preparation and sequencing.
Sequencing libraries were prepared from total RNA samples using Illumina TruSeq RNA Sample Preparation Kit v2 following standard protocols (supplemental Methods, available on the Blood Web site).
Demultiplexing, mapping, and filtering of mouse reads and generation of an expression matrix.
The raw sequencing reads from a lane were demultiplexed with CASAVA 1.8 (Illumina), and aligned to the human (hg19) and mouse (mm10) reference genomes using the TopHat aligner (version 2.0.4).35 Reads aligned to the mouse genome were filtered out by a custom Perl script. After removing the aligned murine reads, sequences aligned to the human genome were used to generate an expression matrix with cufflinks (version 2.0.2) and the hg19 genes gene-transfer-format file from the University of California, Santa Cruz.
Gene set enrichment analysis and visualization
Gene set enrichment analysis was performed as previously described36 using recently reported ABC- and GCB-signature gene sets.37 Gene expression matrices and nonsynonymous mutations were visualized using GENE-E (http://www.broadinstitute.org/cancer/software/GENE-E/).
Whole exome sequencing (WES)
Library preparation and sequencing.
Libraries were prepared as previously described38 using genomic DNAs from the 9 primary LBCLs, 9 tumor specimens from the associated PDX models, 1 NSG mouse (germline), and 2 human (Centre d’Etude du Polymorphisme Humain; Utah residents with ancestry from northern and western Europe; http://hapmap.ncbi.nlm.nih.gov/citinghapmap.html.en) normal, and then sequenced (supplemental Methods).
Demultiplexing, mapping, and SNV, indel, and rearrangement calling.
Samples sequenced in the same lane were demultiplexed using the Picard tools. Read pairs were aligned to the hg19 reference sequence using the Burrows-Wheeler Aligner,39 and data were sorted and duplicate-marked using Picard. Bias in base-quality score assignments due to flow cell, lane, dinucleotide context, and machine cycle were analyzed and recalibrated, and local realignment around insertions or deletions (indels) was performed using the Genome Analysis Toolkit.40,41 Mutation analysis for single-nucleotide variants (SNVs) was performed using MuTect v1.1.4,42 and indel calling was performed using the Genome Analysis Toolkit SomaticIndelDetector tool. SNV and indel calls were further filtered against variants identified in the mouse control sample to remove calls originating from residual mouse DNA in the PDX tumor specimens. Structural variants were detected using BreaKmer.43 SNVs and indels were annotated using Variant Effect Predictor.44 For the comparison of primary tumors and associated PDX models, mutant allele fractions of previously reported DLBCL SNVs were plotted (supplemental Table 2A19-22 ). This analysis was restricted to variants that were absent in mouse and unpaired human normal samples and had an allele fraction of >0.1 and coverage of at least 30× in at least 1 of the primary tumors or the PDX models.
Additional methods
Detailed descriptions of Epstein-Barr-encoded small RNA (EBER) in situ hybridization, immunoglobulin clonality analysis, immunohistochemistry, cell culture, DNA extraction, preparation of libraries for RNA-seq and WES, assessment of CDKN2A and TP53 CNAs, generation of single–tumor cell suspensions, detection of cell surface immunoglobulin expression, and assessment of proliferation and HRK and BCL2A1 transcript abundance after chemical SYK inhibition are included in the supplemental Methods.
Results
Establishment of 9 LBCL PDX models
To develop the PDX models, fresh tumor biopsy samples from 28 primary LBCLs were implanted under the renal capsule of immunocompromised NSG mice. After expansion in vivo, xenotransplanted lymphomas were serially transplanted in additional cohorts of NSG mice. With this approach, 9 of 28 (32%) of the primary LBCLs were propagated for ≥5 generations and considered to be stable LBCL PDX models (Figure 1A). In 5 of 9 PDX models, mice with palpable implanted tumors were found to have pulmonary metastases when euthanized.

Establishment of 9 LBCL PDX models. (A) Hematoxylin and eosin stains of 9 LBCL PDX models propagated under the renal capsule of NSG mice. Original magnification ×200. (B) Epstein-Barr-encoded RNA in situ hybridization of all 9 LBCL PDX models. An EBV-positive classical Hodgkin lymphoma (cHL) served as positive control. Bars represent 100 μm. (C) Clonality of the 9 PDX models assessed and interpreted according to the EuroClonality/BIOMED-2 guidelines.45 An IgH VH-JH spanning PCR (FR2, left) was performed on all 9 LBCL PDX models. Tumors without a clonal peak (LTL-005, LTL-037, and LTL-048) or an inconclusive result (LTL-037) were sequentially analyzed with a second PCR (FR1, right). Genomic DNA (gDNA) of NSG mouse tail served as a negative control, gDNA of peripheral blood mononuclear cells from a healthy human volunteer served as a polyclonal control, and gDNA of the Burkitt lymphoma cell line (BL30) served as a monoclonal control. EBV, Epstein-Barr virus; FR, fragment; PCR, polymerase chain reaction.
Establishment of 9 LBCL PDX models. (A) Hematoxylin and eosin stains of 9 LBCL PDX models propagated under the renal capsule of NSG mice. Original magnification ×200. (B) Epstein-Barr-encoded RNA in situ hybridization of all 9 LBCL PDX models. An EBV-positive classical Hodgkin lymphoma (cHL) served as positive control. Bars represent 100 μm. (C) Clonality of the 9 PDX models assessed and interpreted according to the EuroClonality/BIOMED-2 guidelines.45 An IgH VH-JH spanning PCR (FR2, left) was performed on all 9 LBCL PDX models. Tumors without a clonal peak (LTL-005, LTL-037, and LTL-048) or an inconclusive result (LTL-037) were sequentially analyzed with a second PCR (FR1, right). Genomic DNA (gDNA) of NSG mouse tail served as a negative control, gDNA of peripheral blood mononuclear cells from a healthy human volunteer served as a polyclonal control, and gDNA of the Burkitt lymphoma cell line (BL30) served as a monoclonal control. EBV, Epstein-Barr virus; FR, fragment; PCR, polymerase chain reaction.
To rule out expansion of an EBV-infected cell population, we assessed the EBV status of the subrenal tumor from each PDX model using EBER in situ hybridization. All established PDX models were EBV negative (Figure 1B).
In addition, we assessed the B-cell origin and clonality of each PDX model with a PCR assay targeting the VH-JH region of the heavy chain immunoglobulin locus (Figure 1C).45 Models LTL-013, -014, -025, -026, -030, and -034 were confirmed to be clonal with the IgH VH-JH FR2 PCR (Figure 1C, left); models LTL-005 and -048 were found to be clonal and model LTL-037 was found to be biclonal with the IgH VH-JH FR1 PCR assay (Figure 1C, right).
IHC characterization of the LBCL PDX models
After establishing 9 clonal EBV− LBCL PDX models, each tumor was evaluated with a panel of immunohistochemical (IHC) markers (hematoxylin and eosin, CD20, CD10, MUM1 (IRF4), BCL6, BLC2, and MYC) (Figure 2; Table 1). The morphology and IHC signatures of 8 LBCL PDX models were consistent with the diagnosis of DLBCL (Figure 2A). Each of the DLBCL PDX models had a representative but model-specific pattern of CD10, BCL6, MUM1, BCL2, and MYC expression (Figure 2A; Table 1). All eight DLBCL PDX models were positive for CD20, although LTL-014 expressed CD20 on only 5% of cells. PDX model LTL-037 had 2 populations by CD20 intensity (Figure 2A), consistent with the biclonal IgH analysis (Figure 1C).

IHC characterization of all 9 PDX models. (A) IHC analyses of the indicated markers in all 8 DLBCL PDX models, which were consistent with the diagnosis of DLBCL. (B) IHC assessment of indicated markers in PDX model LTL-048, which is consistent with the diagnosis of PBL. Scale bars, 100 μm. See also Table 1. intracyt., intracytoplasmic.
IHC characterization of all 9 PDX models. (A) IHC analyses of the indicated markers in all 8 DLBCL PDX models, which were consistent with the diagnosis of DLBCL. (B) IHC assessment of indicated markers in PDX model LTL-048, which is consistent with the diagnosis of PBL. Scale bars, 100 μm. See also Table 1. intracyt., intracytoplasmic.
Clonality, and IHC characterization of the LBCL PDX models
| Model ID | Clonality | Diagnosis | IHC, % (intensity) | |||||
|---|---|---|---|---|---|---|---|---|
| CD20 | CD10 | MUM1 | BCL6 | BCL2 | MYC | |||
| LTL-005 | Yes (mono, FR1) | DLBCL | 100 | Negative | 70 (3+) | 60 (2+) | Negative | 40 |
| LTL-013 | Yes (mono, FR2) | DLBCL | 90 | Negative | 10 (3+) | 20 (1+) | 10 (1+) | 80 |
| LTL-014 | Yes (mono, FR2) | DLBCL | 5 | 90 (2+) | 80 (2+) | 30( 2+) | Negative | 99 |
| LTL-025 | Yes (mono, FR2) | DLBCL | 100 | Negative | 100 (3+) | 40 (1+) | 90 (2+) | 90 |
| LTL-026 | Yes (mono, FR2) | DLBCL | 100 | Negative | 50 (3+) | 80 (2+) | 70 (1+) | 90 |
| LTL-030 | Yes (mono, FR2) | DLBCL | 100 | 100 (2+) | Negative | 90 (3+) | 90 (3+) | 10 |
| LTL-034 | Yes (mono, FR2) | DLBCL | 100 | Negative | 100 (3+) | 50 (1+) | 9 (1+) | 70 |
| LTL-037 | Yes (bl, FR1) | DLBCL | 70* | Negative | 70 (3+) | Negative | Negative | 40 |
| LTL-048 | Yes (mono, FR1) | PBL | Negative | 20 (1+) | 100 (3+) | Negative | Negative | 50 |
| Model ID | Clonality | Diagnosis | IHC, % (intensity) | |||||
|---|---|---|---|---|---|---|---|---|
| CD20 | CD10 | MUM1 | BCL6 | BCL2 | MYC | |||
| LTL-005 | Yes (mono, FR1) | DLBCL | 100 | Negative | 70 (3+) | 60 (2+) | Negative | 40 |
| LTL-013 | Yes (mono, FR2) | DLBCL | 90 | Negative | 10 (3+) | 20 (1+) | 10 (1+) | 80 |
| LTL-014 | Yes (mono, FR2) | DLBCL | 5 | 90 (2+) | 80 (2+) | 30( 2+) | Negative | 99 |
| LTL-025 | Yes (mono, FR2) | DLBCL | 100 | Negative | 100 (3+) | 40 (1+) | 90 (2+) | 90 |
| LTL-026 | Yes (mono, FR2) | DLBCL | 100 | Negative | 50 (3+) | 80 (2+) | 70 (1+) | 90 |
| LTL-030 | Yes (mono, FR2) | DLBCL | 100 | 100 (2+) | Negative | 90 (3+) | 90 (3+) | 10 |
| LTL-034 | Yes (mono, FR2) | DLBCL | 100 | Negative | 100 (3+) | 50 (1+) | 9 (1+) | 70 |
| LTL-037 | Yes (bl, FR1) | DLBCL | 70* | Negative | 70 (3+) | Negative | Negative | 40 |
| LTL-048 | Yes (mono, FR1) | PBL | Negative | 20 (1+) | 100 (3+) | Negative | Negative | 50 |
Two populations by CD20 intensity.
Transcriptional heterogeneity within the LBCL PDX models
We next sought to classify the LBCL PDX models with respect to the COO and CCC transcriptional subtypes.2,3 All LBCL PDX models were subjected to RNA-seq. We hypothesized that the infiltrating murine cells in the PDX tumors (Figure 1A) might interfere with RNA-based classification models, especially because RNA-seq protocols select for poly(A)-containing transcripts that are highly conserved between mouse and human. For this reason, we first estimated the percentage of murine transcripts in each PDX model by counting the reads aligned to the mouse reference genome (5.6%-37% of all aligned sequences) (supplemental Figure 1). We then removed the reads aligned to the mouse genome in each model before generating the expression matrix and classifying the PDX models into COO and CCC subtypes.2,3,47
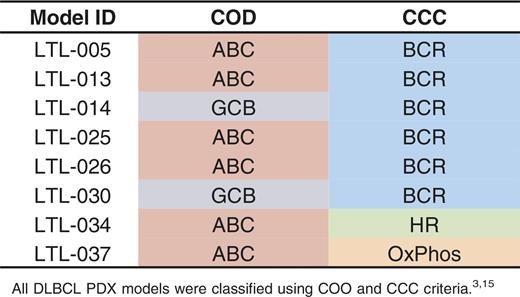
With this approach, of the 8 DLBCL PDX models, 6 were classified as ABC type and 2 were defined as GCB type (Table 2; Figure 3A). Consistent with their COO transcriptional signatures, the 6 ABC-type PDX models expressed increased MUM1, and the 2 GCB tumors had higher CD10 protein levels by IHC (Figure 2A). To further characterize the predicted GCB- and ABC-type DLBCL PDX models, we performed gene set enrichment analyses using recently reported ABC- and GCB-signature gene sets37 (Figure 3B). The respective signatures were significantly enriched in the predicted PDX models (ABC signature in ABC-type PDX models, P < .001; GCB signature in GCB-type PDX models, P = .01; Figure 3B). The PBL PDX model did not exhibit selective expression of specific COO markers, consistent with its unique biology (Figure 3A). Using the CCC classification, 6 of 8 DLBCL PDX models (both of the GCB-type DLBCLs and 4 of 6 ABC-type DLBCLs) were classified as BCR subtype, whereas the remaining 2 PDX models were of non-BCR type (Table 2).

Molecular heterogeneity of LBCL PDX models. (A) Heatmap of the relative expression of ABC- and GCB-signature genes37 in ABC- and GCB-type DLBCL PDX models and the additional PBL PDX model. Note that reads aligned to the mouse genome were filtered before the DLBCL PDX models were classified by COO.3 (B) Gene set enrichment analyses of the ABC-signature genes (upper) and GCB-signature genes (lower) in the ABC- and GCB-type DLBCL PDX models. (C) Detected IGH-MYC (upper) and IGH-BCL2 (lower) translocations in LTL-014 and LTL-030, respectively. Translocations are plotted in their genomic context. Exons are visualized as boxes, with ATG-containing exons in red, coding regions underlined in green, and enhancer regions underlined in black. Numbers of supporting reads (split reads, read pairs) are indicated above, and individual supporting reads are shown below. (D) Protein-perturbing mutations with an allele fraction >0.1 in the LBCL models. Mutations in most frequently reported recurrently mutated genes in primary DLBCL (supplemental Table 2C) are visualized as a color-coded heatmap (red, missense mutation; green, frameshift mutation; purple, nonsense mutation; brown, splice site mutation; white, mutation absent); CNAs in TP53 and CDKN2A are represented as a color-coded heatmap (dark blue, biallelic loss; light blue, monoallelic loss; white, no loss). COO transcriptional subtypes and identified translocations juxtaposing either BCL2 or MYC to the IgH locus are indicated in the legend above of the heatmap. ES, enrichment score.
Molecular heterogeneity of LBCL PDX models. (A) Heatmap of the relative expression of ABC- and GCB-signature genes37 in ABC- and GCB-type DLBCL PDX models and the additional PBL PDX model. Note that reads aligned to the mouse genome were filtered before the DLBCL PDX models were classified by COO.3 (B) Gene set enrichment analyses of the ABC-signature genes (upper) and GCB-signature genes (lower) in the ABC- and GCB-type DLBCL PDX models. (C) Detected IGH-MYC (upper) and IGH-BCL2 (lower) translocations in LTL-014 and LTL-030, respectively. Translocations are plotted in their genomic context. Exons are visualized as boxes, with ATG-containing exons in red, coding regions underlined in green, and enhancer regions underlined in black. Numbers of supporting reads (split reads, read pairs) are indicated above, and individual supporting reads are shown below. (D) Protein-perturbing mutations with an allele fraction >0.1 in the LBCL models. Mutations in most frequently reported recurrently mutated genes in primary DLBCL (supplemental Table 2C) are visualized as a color-coded heatmap (red, missense mutation; green, frameshift mutation; purple, nonsense mutation; brown, splice site mutation; white, mutation absent); CNAs in TP53 and CDKN2A are represented as a color-coded heatmap (dark blue, biallelic loss; light blue, monoallelic loss; white, no loss). COO transcriptional subtypes and identified translocations juxtaposing either BCL2 or MYC to the IgH locus are indicated in the legend above of the heatmap. ES, enrichment score.
Genetic characterization of LBCL PDX models
To gain insight into the genetic alterations in each LBCL PDX model, we subjected PDX tumor specimens and an NSG mouse germline control to WES. The classical WES bait set was extended with baits spanning 39 chromosomal regions that are rearranged in specific hematologic malignancies (supplemental Table 1A). We first estimated the murine contribution to the LBCL PDX WES data and determined the median mouse sequences to be ∼3.4% of total reads. There were relatively fewer murine reads in the WES data than in the RNA-seq data, likely due to the specific design of the WES bait set.
Chromosomal rearrangements in the LBCL PDX models.
To detect chromosomal rearrangements, the DNA-sequencing data (WES plus custom bait set) for each PDX model was analyzed with the BreaKmer algorithm.43 Chromosomal translocations were detected in 2 of the PDX models: t(8;14)/IgH-MYC in LTL-014 and t(14;18)/IgH-BCL2 in LTL-030 (Figure 3C; supplemental Table 1B). In both cases, the chromosomal translocations juxtaposed either the BCL2 or the MYC locus to strong IgH enhancer elements (Figure 3C). Of note, the breakpoint in the t(14;18)/IgH-BCL2 translocation places the IgH enhancer ∼6 kb distal to the end of the BCL2 gene between the major and minor breakpoint regions, as previously described in primary tumor specimens.48 Consistent with these genetic alterations, the PDX models with the highest MYC and BCL2 expression were LTL-014 and LTL-030, respectively (Figure 2A). The t(14;18)/IgH-BCL2 translocation occurred in a GCB-type DLBCL PDX model (LTL-030), as reported in primary DLBCL.7 Of interest, the t(8;14)/IgH-MYC translocation occurred in the second GCB-type DLBCL PDX model, suggesting that both GCB-type models required additional genetic drivers for murine engraftment. In primary DLBCLs, BCL2 and MYC translocations are known adverse prognostic features in GCB-type tumors.49-53 We also noted that the DLBCL PDX model with the t(8;14)/IgH-MYC translocation (LTL-014) had only 5% CD20-positive cells (Figure 2; Table 1).
Mutations in the LBCL PDX models.
After applying a rigorous filter to remove common human or mouse single-nucleotide polymorphisms, we next queried for the most frequently reported recurrent mutations in primary DLBCLs (mutations identified as significant in 2/4 publications19-22 ; supplemental Table 2). Mutations present in at least 1 DLBCL PDX model are included in Figure 3D and supplemental Table 2C.
One of the GCB-type DLBCL PDX models, LTL-030, had alterations of GNA13, CREBBP, and EZH2, all previously linked to the GCB subtype.9,20,21 ABC-type DLBCL PDX models had a larger spectrum of mutations, including MYD88 in association with CARD11 and CD79B (LTL-013), PIM1 (LTL-005), or PRDM1 (LTL-034), or CD79B with additional alterations (LTL-026), as previously reported in primary ABC-type DLBCLs.10,12,20,21 Certain PDX models had additional mutations, including B2M, MLL2, MEF2B, NOTCH1, and TP53.16,17,20,21,54 None of the 37 most recurrently mutated genes in DLBCL was identified in the PBL PDX model.
Importantly, we found that only 25% (2/8) of the DLBCL PDX models harbored inactivating TP53 mutations (Figure 3D). This finding is consistent with the reported incidence of TP53 mutations in primary DLBCLs15,19-22,55 and unlike the near-uniform presence of TP53 somatic mutations in DLBCL cell lines.15
CNAs of TP53 and CDKN2A.
We previously reported that primary DLBCLs often exhibit CNAs of TP53 or additional modifiers of the p53 pathway in addition to less frequent somatic mutations of TP53.15 For this reason, we used a targeted real-time PCR assay to assess copy numbers of TP53 and its upstream modifier, CDKN2A, in the PDX models (Figure 3D). Five of the 8 DLBCL PDX models (LTL-005, LTL-013, LTL-014, LTL-025, and LTL-026) exhibited single copy loss of TP53. In LTL-013 and LTL-014, this resulted in functional biallelic inactivation of TP53 because the second TP53 allele was inactivated by somatic mutation (Figure 3D). Because the CDKN2A locus encodes 2 alternative transcripts, p16INK4A and p19ARF, we assessed the CDKN2A locus with 3 separate copy number assays that covered the representative exons in each transcript. Two of the PDX models (LTL-025 and LTL-034) had complete loss of all coding exons of CDKN2A, and an additional model (LTL-005) lost both copies of the p16INK4A-encoding portion of the gene. Two additional PDX models (LTL-026 and LTL-014) had single copy loss of the coding CDKN2A exons. Overall, we detected CNAs of TP53 or CDKN2A in 75% (6/8) of the DLBCL PDX models and detected somatic TP53 mutations in only 25% (2/8) of models, similar to the reported incidence of p53 pathway alterations in primary DLBCLs.15
Genetic stability of PDX models.
To evaluate the genetic stability of the PDX models, we compared genetic alterations in each primary LBCL with its associated PDX model. We employed WES with the same extended bait set to capture all mutations perturbing the protein sequence and selected chromosomal rearrangements. The identical chromosomal rearrangements of IgH/MYC (LTL-14) or IgH/BCL2 (LTL-030) were detected at base-pair resolution in the primary tumors and associated PDX models (supplemental Figure 2).
We also compared the mutant allele fraction (frequency of a mutation at a particular locus) in each of the primary tumors and associated PDX models (Figure 4; supplemental Table 2A19-22 ). The majority of DLBCL SNVs were present at similar mutant allele fractions in the PDX models and the original primary tumors (Figure 4). For example, the mutant allele fractions of almost all DLBCL SNVs were comparable in LTL-005, LTL-013, LTL-026, and LTL-037 primary tumors and PDX models. In certain models, select DLBCL SNVs were more common in the PDX model than in the primary tumor (TP53 in LTL-014, KMT2D [MLL] and TMEM30A in LTL-025, B2M and MEF2B in LTL-030, and PRDM1 [BLIMP1] in LTL-034). Mutant allele fractions of hallmark DLBCL SNVs, including MYD88, PIM1, CARD11 and CD79B, TNFAIP3, EZH2, and POUZF2, were similar in the primary tumors and PDX models. Of note, 2 common DLBCL SNVs were detected in the primary tumors but not in the PDX models (GNA13 in LTL-014 and TP53 in LTL-037). These data provide further evidence that the PDX models largely capture and retain the genetic heterogeneity of the primary DLBCLs.

Comparison of mutant allele fraction between primary tumor and associated PDX model. We plotted the mutant allele fraction (frequency of a mutation at a particular locus) in the primary tumors (x-axis) and the associated PDX models (y-axis). SNVs in genes reported to be mutated in at least 1 of 4 DLBCL sequencing series are represented in gray, and SNVs in genes reported to be mutated in at least 2 of 4 DLBCL sequencing series are labeled in red (supplemental Table 2A19-22 ). Multiple SNVs in the same gene are noted (PIM1 in LTL-005, KMT2D in LTL-025, and POU2F2 in LTL-034). Mutant allele fractions along the diagonal (x = y, identical mutant allele fractions, dotted line) indicate similar clonal frequencies in the primary tumor and associated PDX model. Mutant alleles that are more abundant in the PDX model than in the primary tumors are above the diagonal. Allele fractions ±0.2 from the identical mutant allele fractions are visualized in gray.
Comparison of mutant allele fraction between primary tumor and associated PDX model. We plotted the mutant allele fraction (frequency of a mutation at a particular locus) in the primary tumors (x-axis) and the associated PDX models (y-axis). SNVs in genes reported to be mutated in at least 1 of 4 DLBCL sequencing series are represented in gray, and SNVs in genes reported to be mutated in at least 2 of 4 DLBCL sequencing series are labeled in red (supplemental Table 2A19-22 ). Multiple SNVs in the same gene are noted (PIM1 in LTL-005, KMT2D in LTL-025, and POU2F2 in LTL-034). Mutant allele fractions along the diagonal (x = y, identical mutant allele fractions, dotted line) indicate similar clonal frequencies in the primary tumor and associated PDX model. Mutant alleles that are more abundant in the PDX model than in the primary tumors are above the diagonal. Allele fractions ±0.2 from the identical mutant allele fractions are visualized in gray.
Assessing integrity of BCR signaling in PDX models
After establishing and genetically characterizing the 9 LBCL PDX models, we assessed their utility for functional analyses of BCR signaling. To this end, we excised subrenal tumor from each of the PDX models, generated viable tumor cell suspensions, and assessed surface immunoglobulin expression by flow cytometry (Figure 5A). Six of the 8 DLBCL PDX models had expression of surface IgM, suggesting that these models derived from non-class-switched GCBs (Figure 5A). The remaining 2 DLBCL PDX models and the PBL PDX model had no surface IgM expression; none of the PDX models had surface IgG expression (Figure 5A). Of interest, all of the surface immunoglobulin–positive LBCL PDX models were classified as BCR-type DLBCLs by CCC.5 These surface immunoglobulin–positive BCR-type DLBCLs belonged to both COO subtypes (GCB-type: LTL-014 and LTL-30; ABC-type: LTL-005, LTL-013, LTL-025, and LTL-026; Figure 5A).

Analyses of cell surface immunoglobulin and BCR signaling in the LBCL PDX models. (A) Single-cell suspensions from each LBCL PDX model were gated for human CD45-positive cells and analyzed for surface immunoglobulin (IgG, red; IgM, orange; isotype, gray). CCC and COO subtypes of each model are indicated above the flow histograms. (B) Cellular proliferation of PDX tumor cell suspensions after chemical SYK inhibition with entospletinib (GS-9973) for 24 hours. (C,D) HRK (C) and BCL2A1 (D) transcript abundance following entospletinib (GS-9973) treatment. Experiments were performed in triplicate. A representative experiment of biological duplicates in shown. DMSO, dimethylsulfoxide. The P values were obtained using a Student t test (*P < .05).
Analyses of cell surface immunoglobulin and BCR signaling in the LBCL PDX models. (A) Single-cell suspensions from each LBCL PDX model were gated for human CD45-positive cells and analyzed for surface immunoglobulin (IgG, red; IgM, orange; isotype, gray). CCC and COO subtypes of each model are indicated above the flow histograms. (B) Cellular proliferation of PDX tumor cell suspensions after chemical SYK inhibition with entospletinib (GS-9973) for 24 hours. (C,D) HRK (C) and BCL2A1 (D) transcript abundance following entospletinib (GS-9973) treatment. Experiments were performed in triplicate. A representative experiment of biological duplicates in shown. DMSO, dimethylsulfoxide. The P values were obtained using a Student t test (*P < .05).
Next, we assessed the functional integrity of BCR signaling in the panel of LBCL PDX models using the selective SYK inhibitor entospletinib (GS-9973), which is currently in clinical trials for multiple B-cell malignancies.56-58 Like other chemical SYK inhibitors and molecular SYK depletion,5 single-agent entospletinib (GS-9973) selectively blocked the proliferation of BCR-dependent DLBCL cell lines, abrogated the phosphorylation of SYK (pSYK525/526) and downstream pathway components, and modulated the expression of specific BCL2 family members (HRK and BCL2A1) in BCR-dependent DLBCL cell lines with low- and high baseline NF-κB activity (supplemental Figure 3).
In single-cell suspensions of the LBCL PDX tumors, entospletinib treatment significantly decreased the proliferation of all 6 BCR-type DLBCLs but had no effect on the non-BCR-type DLBCLs or the PBL (Figure 5B). Given the distinctive SYK/PI3K-dependent signaling and survival pathways in DLBCLs with low or high baseline NF-κB, which largely correspond to BCR-dependent GCB- or ABC-type DLBCLs, respectively,5 we next assessed readouts of these pathways (Figure 5C-D). As in DLBCL cell lines (supplemental Figure 3) and primary DLBCL patient samples,5 chemical SYK inhibition selectively induced the proapoptotic BH3-only family member HRK in both of the BCR-dependent GCB-type DLBCL PDX samples (Figure 5C) and decreased expression of the antiapoptotic BCL2-family member BCL2A1 in BCR-dependent ABC-type DLBCL PDX tumors (Figure 5D). These data indicate that the molecularly characterized DLBCL PDX models faithfully reflect defined differences in BCR dependence, downstream signaling, and survival pathways.
Discussion
We have generated and characterized a panel of LBCL PDX models, including 8 that reflect the immunophenotypic, transcriptional, genetic, and functional heterogeneity of primary DLBCL. One additional model is of the immunophenotypically and morphologically distinct LBCL subtype, PBL.45 These data indicate that implanting fresh LBCL specimens within the highly vascularized subrenal capsule of immunodeficient mice is feasible and effective, as reported for certain solid tumors.33,34
Our genomic characterization of human PDX models revealed the importance of computationally subtracting murine reads in RNA-seq data. RNA-seq data contained a higher percentage of murine reads than DNA-sequencing data (WES), likely due to the associated differences in library preparation. For these reasons, we eliminated mouse-specific reads from our RNA-seq data before characterizing the transcriptional signatures of the LBCL PDX models and used the DNA-seq data to call mutations and chromosomal rearrangements. These approaches to the analysis of transcriptional signatures and mutation calling will be broadly applicable across tumor types.
Applying the RNA-based COO classification to the DLBCL PDX series, 6 of 8 models were classified as ABC type and 2 as GCB type. The higher engraftment rate of ABC-type DLBCL PDX models may reflect the more aggressive course of the clinical disease.59 Of interest, 5 of 6 ABC-type PDX models had additional genetic alterations of p53/cell cycle pathway components, including 3 models with biallelic loss of CDKN2A, 1 model with biallelic inactivation of TP53 (mutation and single copy loss), and another model with single copy loss of TP53. Furthermore, the 2 GCB-type PDX models had additional adverse genetic features: t(14;18)/IgH-BCL2 translocation or t(8;14)/IgH-MYC translocation.49-53 The GCB-type PDX model with the MYC translocation also had biallelic inactivation of TP53 (mutation and single copy loss). In this initial series of DLBCL PDX models, tumors with more aggressive genetic features are overrepresented, suggesting that such lymphomas may engraft more readily in NSG mice. These findings are consistent with those in solid tumors in which engraftment and PDX generation were adversely associated with patient overall survival.27,60
By performing WES of the DLBCL PDX models, we identified mutations associated with COO subtype (ABC: MYD88, CD79B, CARD11, and PIM110,12,21 ; GCB: GNA13, EZH2, and CREBBP8,9,61 ) and additional reported alterations (B2M, MLL2, TNFAIP3, and MEF2B).6,15-17 Only 25% (2/8) of the DLBCL PDX models harbored inactivating TP53 mutations, whereas 75% (6/8) of tumors exhibited CNAs of TP53 or its upstream modifier, CDKN2A. These data are consistent with the reported incidence and type of TP53 alterations in primary DLBCLs and contrast sharply with the near-uniform presence of inactivating TP53 mutations in DLBCL cell lines.15 For these reasons, the DLBCL PDX panel maybe particularly useful for the analysis of p53 mimetics62 or MDM2/MDM4 inhibitors63 which require wild-type p53 activity.
By comparing the mutant allele fraction in primary LBCLs and the associated PDX models, we were able to assess the stability of the PDX genetic signature. Most of the LBCL mutations were present at similar allele fractions in the PDX models and the associated primary tumors, indicating that these models largely retain the complex genetic signature of primary LBCLs.
As noted, 6 of the DLBCL PDX models were identified as BCR type based on CCC transcriptional profiles, including 4 ABC-type and 2 GCB-type tumors. The BCR-type DLBCLs were selectively surface IgM positive and sensitive to chemical inhibition of SYK using entospletinib. As in our previous studies,5 the readouts of SYK inhibition differed in BCR-dependent ABC- and GCB-type DLBCL PDX models: downregulation of NF-κB targets such as BCL2A1 in ABC-type tumors vs HRK upregulation in GCB-type tumors. These studies highlight the utility of having a diverse and well-characterized DLBCL PDX panel for evaluating targeted inhibitors of subtype-specific survival programs. In addition, these studies illustrate the usefulness of PDX models as a renewable source of viable tumor cells for the analysis of sensitivity and resistance to novel targeted agents.
In summary, we have established a molecularly characterized and faithful panel of PDX models of primary DLBCL and PBL and demonstrated their usefulness in evaluating proximal BCR pathway inhibition. We anticipate that the panel of LBCL PDX models will facilitate rational target identification and preclinical drug discovery. In addition, the associated insights regarding both the establishment and the molecular characterization of the LBCL PDX models will be broadly applicable to other tumor types.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the Claudia Adams Barr Program in Innovative Basic Cancer Research (B.C.), the Terry Fox Research Institute Program Project Grant #1023 (R.D.G.), National Institutes of Health National Cancer Institute grant (P01 CA092625) (M.A.S.) and a Leukemia & Lymphoma Society Translational Research Program award (M.A.S.).
Authorship
Contribution: B.C. designed and performed the research, analyzed the data, and wrote the paper; H.C., A.W., S.J.R., and Y.W. designed and performed the research and analyzed the data; M.D.D., D.G., L.C., M.G.M.R., J.O., L.Z., Y.T., R.P.A., A.R.T., G.S.P., S.M., and G.G.W. performed the research and analyzed the data; A.L.C., F.v.B., P.F., and H.H.S. performed the research; P.v.H., J.C.A., R.D.G., and D.M.W. designed the research; and M.A.S. designed the research, analyzed the data, and wrote the paper.
Conflict-of-interest disclosure: D.M.W. received consulting fees and research support from Novartis. M.A.S. has served on advisory boards for Gilead Sciences. The remaining authors declare no competing financial interests.
Correspondence: Margaret Shipp, Dana-Farber Cancer Institute, Harvard Medical School, 450 Brookline Ave, Mayer 513, Boston, MA 02215; e-mail: margaret_shipp@dfci.harvard.edu.
