

Key Points
RGS18 acts as a brake on persistent or inappropriate platelet activation after it is released from binding sites in resting platelets.
Control of free RGS18 levels provides a mechanism for coordinating signaling networks in platelets.
Abstract
Most platelet agonists activate platelets by binding to G-protein–coupled receptors. We have shown previously that a critical node in the G-protein signaling network in platelets is formed by a scaffold protein, spinophilin (SPL), the tyrosine phosphatase, Src homology region 2 domain-containing phosphatase-1 (SHP-1), and the regulator of G-protein signaling family member, RGS18. Here, we asked whether SPL and other RGS18 binding proteins such as 14-3-3γ regulate platelet reactivity by sequestering RGS18 and, if so, how this is accomplished. The results show that, in resting platelets, free RGS18 levels are relatively low, increasing when platelets are activated by thrombin. Free RGS18 levels also rise when platelets are rendered resistant to activation by exposure to prostaglandin I2 (PGI2) or forskolin, both of which increase platelet cyclic adenosine monophosphate (cAMP) levels. However, the mechanism for raising free RGS18 is different in these 2 settings. Whereas thrombin activates SHP-1 and causes dephosphorylation of SPL tyrosine residues, PGI2 and forskolin cause phosphorylation of SPL Ser94 without reducing tyrosine phosphorylation. Substituting alanine for Ser94 blocks cAMP-induced dissociation of the SPL/RGS/SHP-1 complex. Replacing Ser94 with aspartate prevents formation of the complex and produces a loss-of-function phenotype when expressed in mouse platelets. Together with the defect in platelet function we previously observed in SPL−/− mice, these data show that (1) regulated sequestration and release of RGS18 by intracellular binding proteins provides a mechanism for coordinating activating and inhibitory signaling networks in platelets, and (2) differential phosphorylation of SPL tyrosine and serine residues provides a key to understanding both.
Introduction
Circulating blood platelets play a central role in hemostasis and thrombosis. Key external determinants of the platelet activation state include the local concentration of platelet agonists, the presence of inhibitors of thrombin production and activity, and the local production by endothelial cells of prostaglandin I2 (PGI2) and nitric oxide, both of which raise cyclic nucleotide levels in platelets. Many platelet agonists, including thrombin, adenosine 5′-diphosphate (ADP), and thromboxane A2 (TxA2), activate platelets via G proteins and G-protein–coupled receptors.1 Whereas agonists favor platelet activation, increasing cyclic adenosine monophosphate (cAMP) levels inhibit platelet reactivity, rendering them resistant to activation by any of the agonists that they subsequently encounter. Thus, drugs that raise platelet cAMP levels have clinically useful antiplatelet activity2,3 and, conversely, knocking out platelet PGI2 receptors in mice produces a prothrombotic phenotype.4
It is within this context of opposing influences on platelet activation that we have considered the role of the regulators of G-protein signaling (RGS) proteins that are expressed in platelets. Although agonist-occupied G-protein–coupled receptors initiate signaling by promoting the exchange of guanosine diphosphate (GDP) for guanosine triphosphate (GTP) on G-protein α subunits, RGS proteins terminate G-protein signaling by accelerating the hydrolysis of Gα-bound GTP, allowing Gα to reassociate with Gβγ.5-7 There are at least 37 genes encoding RGS proteins in the human genome. At least 8 have been detected at the transcript level in platelets, but protein studies suggest that human and mouse platelets express predominantly RGS10 and RGS18.8-11 Both proteins are relatively small, consisting primarily of a characteristic RGS domain that interacts with Gα.12 Each can serve as GTPase-accelerating proteins for Giα and Gqα, but not Gsα.13-17 RGS18 is primarily expressed in hematopoietic cells14,16,18-20 whereas RGS10 is widely expressed.21-23 In addition to being expressed in platelets, there is a small, but growing, body of evidence that RGS proteins are biologically relevant regulators of platelet activation. Thus, it has been shown that platelet reactivity increases in mice expressing a Gi2α variant incapable of binding RGS proteins as a class,24 as does platelet reactivity in mice lacking RGS18.25
If RGS proteins are to be considered to be brakes on platelet activation, then a question arises about the timing of the application of those brakes so that platelets can be activated when needed, but only when necessary. In other words, is there a separate mechanism for regulating the interaction of RGS proteins with activated G proteins in platelets? Again, previous observations at least suggest that this may be the case. RGS10 and RGS18 have both been shown to bind to the scaffold protein, spinophilin (neurabin-II or SPL) in platelets8 and the 14-3-3 family member, 14-3-3γ, has been shown to bind to RGS18.26 The mechanism that governs these interactions and the timing of the association and dissociation of the RGS proteins with each of these partners is different. We have shown that, in resting platelets, RGS10 and RGS18 form a complex with SPL that includes the tyrosine phosphatase, Src homology region 2 domain-containing phosphatase-1 (SHP-1). Within this complex, SPL is phosphorylated on tyrosine residues Y398 and Y483, with phosphorylated Y398 providing a binding site for SHP-1.8 Platelet activation by thrombin and TxA2, but not by collagen or ADP, activates SHP-1, triggers dephosphorylation of SPL, and causes gradual, but complete, dissociation of the SPL/RGS/SHP-1 complex. Gegenbauer and colleagues26 have studied the interaction of RGS18 with 14-3-3γ and shown that binding occurs in resting platelets and increases when platelets are activated. 14-3-3 proteins bind phosphorylated serine residues. Binding in this case is mediated by phosphorylated Ser49 and Ser218 in RGS18. Phosphorylation of Ser49 increases when platelets are activated. Binding to 14-3-3γ inhibits the interaction of RGS18 with activated G proteins.27 Notably, cAMP-dependent phosphorylation of RGS18 on Ser216 when platelets were incubated with PGI2 caused it to dissociate from 14-3-3γ.27
These observations suggest a model in which RGS10 and RGS18 provide a brake on excessive platelet activation, with the availability of the RGS proteins subject to divergent interactions with SPL and 14-3-3γ. Studies on SPL−/− mice are consistent with this hypothesis: we found that platelet function is reduced in the absence of SPL, as if loss of this binding protein results in an increase in available RGS proteins.8
Here, we have tested and extended this model, focusing on RGS18 and asking, first, whether there is a bona fide increase in free RGS18 levels when resting platelets are activated and, second, how such an increase is coordinated in different states of platelet reactivity. Answering the first question required the development of an assay for free RGS18 in platelets. Answering the second led to a comparison between resting platelets, activated platelets, and platelets rendered resistant to activation by exposure to PGI2. The data demonstrate an increase in available (“free”) RGS18 in platelets incubated either with thrombin or PGI2. Notably, the mechanism that triggers the release of RGS18 from SPL by PGI2 turns out to be distinct from both the mechanism that underlies thrombin-induced dissociation of the SPL/RGS/SHP-1 and the mechanism by which an increase in cAMP levels separates RGS18 from 14-3-3γ.
Methods and materials
Materials
Prostacyclin (PGI2) and forskolin were purchased from Sigma-Aldrich; thrombin was obtained from Haematologic Technologies, Inc. Goat anti-SPL (A-20) was purchased from Santa Cruz Biotechnology; the phosphotyrosine antibody was obtained from EMD Millipore. Rabbit anti-Flag and mouse (9B11) and rabbit (71D10) anti-Myc were obtained from Cell Signaling Technology. Mouse anti-Flag (M2) was obtained from Sigma-Aldrich. pEx39Not+ containing the complementary DNA encoding rat SPL was a gift from Dr Patrick Allen (Yale University). Full-length SPL was subcloned into the pCR-Blunt II-TOPO vector using the Zero Blunt TOPO PCR cloning kit (Invitrogen). SPL was additionally subcloned into the pCMV-3B expression vector, using restriction sites for HindIII and SalI incorporated by synthetic oligonucleotides. Long-term stable production of retrovirus (LZRS)–SPL-green fluorescent protein (GFP) plasmids were kindly provided by Dr Ona Bloom (The Feinstein Institute for Medical Research). Gi2α plasmids were purchased from University of Missouri-Rolla (UMR, now Missouri University of Science and Technology) cDNA Resource Center (Rolla, MO). Gi2α was then subcloned into the pGEX-4T expression vector. Chinese hamster ovary (CHO) cells and Phoenix cells were purchased from ATCC. SPL−/− mice and the pSer94-specific SPL antibody were generously provided by Dr Paul Greengard (Rockefeller University).28 The mice had been backcrossed into C57 Bl/6 at least 7 times. The studies were carried out with institutional animal care and use committee–approved protocols.
Fetal liver cell transplantation
Fetal liver cells were harvested from SPL−/− embryos at embryonic days 15.5 to 17.5 (E15.5 to E17.5) and mononuclear cells were purified with Lympholyte (Cedarlane Labs) gradient and maintained overnight in Iscove modified Dulbecco medium (Invitrogen) supplemented with 10% fetal bovine serum, 100 ng/mL murine stem cell factor, 20 ng/mL interleukin-3, and 10 ng/mL interleukin-6. The cells were infected twice with SPL retrovirus. A total of 2 × 106 were transplanted by retroorbital injection (200 μL per mouse) into recipient mice (8-10 weeks old) conditioned with a split lethal dose of 1100-rad irradiation. Platelet studies were performed 4 to 6 weeks after transplantation. SPL-GFP expression was detected by flow cytometry (BD FACSCalibur; BD Biosciences).
Platelet aggregation
Blood was taken from the inferior vena cava of anesthetized mice (100 mg/kg Nembutal) using a heparinized syringe (150 U/mL, 1:9 dilution with blood). Blood was diluted 1:1 with N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES)-Tyrode buffer, and spun at 129g for 7 minutes to prepare platelet-rich plasma. Platelet counts (Beckman-Coulter Z1) were adjusted to 2.5 × 108/mL with autologous platelet-poor plasma. Aggregation was observed in a dual-channel Chrono-log lumiaggregometer.
Cytosolic calcium
CHO cells were transfected with Myc-SPL, hemagglutinin (HA)-RGS18, resuspended in phosphate-buffered saline (PBS) containing 1 mM CaCl2 and 0.1% bovine serum albumin, and incubated with 10 μg/mL Fura-2 AM for 30 minutes at 37° in the dark. Cells were then washed twice with PBS/CaCl2 buffer and diluted to 106 cells per mL in PBS/CaCl2 buffer. Changes in cytosolic Ca2+ were detected using an SLM/Aminco model AB2 fluorescence spectrophotometer with excitation at 340 and 380 nm and emission at 510 nm.
Free RGS18 levels
BL21 Escherichia coli cells (Agilent Technologies) were transformed with vector containing pGEX4T-Gi2α. Glutathione S-transferase (GST)-Gi2α fusion protein expression was induced overnight at room temperature with 1 mM isopropyl-β-D-thiogalactopyranoside. GST-Gi2α fusion protein was purified using Glutathione-Sepharose beads from GE Healthcare. Beads were activated with aluminum tetrafluoride (AlF4−; 10 mM NaF and 30 μM AlCl3) and GDP (100 μM) or with GDP (100 μM) alone as a negative control. Platelets were incubated with 15 μM PGI2 or 20 μM forskolin for 10 minutes or activated with 50 μM SFLLRN for 3 minutes at 37°C and then lysed. Lysates were incubated with activated GST-Gi2α–bound beads or control beads overnight. After 3 washes with lysis buffer, bound proteins were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and blotted with an anti-RGS18 antibody.
Statistical analysis
Results were presented as means ± standard error of the mean (SEM) as shown in figure legends, and data were analyzed using the Student t test. P values < .05 were considered to be statistically significant.
For additional materials and methods, see supplemental Methods (see supplemental Data available at the Blood Web site).
Results
Changes in free RGS18 levels in platelets in response to agonists and antagonists of platelet activation
RGS proteins serve as GTPase-accelerating proteins for Gq and Gi family members by binding directly to the GTP-bound α subunits of these proteins.17 To test the hypothesis that there is a change during platelet activation in the amount of “free” RGS18 available to interact with G proteins, we established an assay using a GST-Gi2α fusion protein as bait to retrieve RGS18 from platelet lysates. AlF4− was added to mimic the GTP hydrolysis transition state of Gi2α: that is, the state of Gα that is most avidly bound by RGS domains.13 As shown in Figure 1 and supplemental Figure 1, the amount of RGS18 captured by GST-Gi2α in the presence of AlF4− increased when human platelets were incubated with the protease-activated receptor 1 (PAR1; thrombin receptor) agonist peptide, SFLLRN, or the TxA2 mimetic, U46619. In contrast, there was no increase in captured RGS18 when platelets were incubated with ADP in the presence of aspirin to inhibit TxA2 synthesis. The differences between SFLLRN, U46619, and ADP in this regard mirrors their abilities to cause dissociation of the SPL/RGS18/SHP-1 complex: dissociation of the complex occurs with SFLLRN and U46619, but not with ADP.8

GST-Gi2α pulldown of RGS18 from human platelet lysates. Human platelets. (A) Lysates were prepared from resting platelets and from platelets incubated with forskolin (20 µM), PGI2 (15 µM), or the PAR1 agonist peptide, SFLLRN (50 µM). The lysates were then incubated with GST-Gi2α coupled to glutathione beads in the presence of GDP plus AlF4− or GDP alone as indicated. Bound proteins were subjected to electrophoresis and probed with RGS18 and Gi2α antibodies to detect RGS18 and GST-Gi2α fusion protein, respectively. (B) Summary of 3 experiments expressed as the percentage of the result obtained with resting platelets (mean ± SEM). P values are relative to resting platelets. IB, immunoblotting.
GST-Gi2α pulldown of RGS18 from human platelet lysates. Human platelets. (A) Lysates were prepared from resting platelets and from platelets incubated with forskolin (20 µM), PGI2 (15 µM), or the PAR1 agonist peptide, SFLLRN (50 µM). The lysates were then incubated with GST-Gi2α coupled to glutathione beads in the presence of GDP plus AlF4− or GDP alone as indicated. Bound proteins were subjected to electrophoresis and probed with RGS18 and Gi2α antibodies to detect RGS18 and GST-Gi2α fusion protein, respectively. (B) Summary of 3 experiments expressed as the percentage of the result obtained with resting platelets (mean ± SEM). P values are relative to resting platelets. IB, immunoblotting.
Notably, there was also an increase in free RGS18 when platelets were incubated with either PGI2 or forskolin, both of which cause an increase in platelet cAMP levels and render platelets resistant to the activating effects of agonists. PGI2 binds to platelet prostacyclin (IP) receptors, activating adenylyl cyclase via Gs; forskolin activates adenylyl cyclase directly. The increase in free RGS18 in PGI2- and forskolin-stimulated platelets matched the increase that occurred with SFLLRN (Figure 1; supplemental Figure 1).
These initial results support the previously proposed increase in free RGS18 following dissociation of the SPL/RGS18/SHP-1 complex when platelets are activated by a thrombin receptor agonist,29 but leave unexplained the increase in free RGS18 observed in PGI2- and forskolin-stimulated platelets. To examine the mechanism for that increase, intact platelets were incubated with forskolin or PGI2, RGS18 was immunoprecipitated from platelet lysates, and the precipitates were probed for SPL (Figure 2A-B). The results show that, compared with resting platelets, significantly less SPL coprecipitates with RGS18 in platelets exposed to forskolin or PGI2. Adding okadaic acid to inhibit serine/threonine phosphatases during the incubation with PGI2 and forskolin produced no further effect. There was also a decrease in SHP-1 binding to SPL in platelets incubated with PGI2 and forskolin (Figure 2C-D). Thus, like thrombin and TxA2, both methods of raising platelet cAMP levels (forskolin and PGI2) cause dissociation of the SPL/RGS18/SHP-1 complex. Notably, however, in contrast to thrombin,8 there was no reduction in SPL tyrosine phosphorylation when platelets were incubated with either PGI2 or forskolin (Figure 2E-F).

Forskolin and PGI2 cause dissociation of the SPL/RGS/SHP-1 complex, but do not affect SPL tyrosine phosphorylation. (A) Human platelets were incubated with 20 µM forskolin (Forsk) ± 1 µM okadaic acid as indicated. Proteins were precipitated with anti-RGS18 or nonimmune immunoglobulin (Ig) and then probed with anti-SPL before reprobing with anti-RGS18. The graph shows mean ± SEM. (B) Identical to panel A except 15 µM PGI2 was used instead of forskolin. Panels C and D are identical to panels A and B except that immunoprecipitation was performed with anti-SHP-1 instead of anti-RGS18. Panels A-C, N = 4; panel D, N = 3. (E) Human platelets were incubated with 20 µM forskolin ± 1 µM okadaic acid as indicated. Proteins were precipitated with the anti-phosphotyrosine antibody, 4G10, or nonimmune Ig and then probed with anti-SPL. The graph summarizes 4 experiments (mean ± SEM). Panel F is identical to panel E except 15 µM PGI2 was used instead of forskolin; N = 3. P values are relative to resting platelets.
Forskolin and PGI2 cause dissociation of the SPL/RGS/SHP-1 complex, but do not affect SPL tyrosine phosphorylation. (A) Human platelets were incubated with 20 µM forskolin (Forsk) ± 1 µM okadaic acid as indicated. Proteins were precipitated with anti-RGS18 or nonimmune immunoglobulin (Ig) and then probed with anti-SPL before reprobing with anti-RGS18. The graph shows mean ± SEM. (B) Identical to panel A except 15 µM PGI2 was used instead of forskolin. Panels C and D are identical to panels A and B except that immunoprecipitation was performed with anti-SHP-1 instead of anti-RGS18. Panels A-C, N = 4; panel D, N = 3. (E) Human platelets were incubated with 20 µM forskolin ± 1 µM okadaic acid as indicated. Proteins were precipitated with the anti-phosphotyrosine antibody, 4G10, or nonimmune Ig and then probed with anti-SPL. The graph summarizes 4 experiments (mean ± SEM). Panel F is identical to panel E except 15 µM PGI2 was used instead of forskolin; N = 3. P values are relative to resting platelets.
Mechanism of cAMP-induced dissociation of the SPL/RGS18/SHP-1 complex
Serine 94 in SPL is highly conserved and, based on its surrounding sequence, a potential target for phosphorylation by cAMP-dependent protein kinase A (PKA).30 Western blotting with a phospho-Ser94–specific SPL antibody shows that there is little, if any, detectable SPL pSer94 in resting platelets and in platelets activated with SFLLRN, but a large increase occurs when platelets are incubated with forskolin or PGI2 (Figure 3; supplemental Figure 2).

SPL becomes phosphorylated on serine 94 in platelets incubated with forskolin or PGI2. (A) Human platelets were incubated with 20 µM forskolin (Forsk) ± 1 µM okadaic acid (OA) as indicated. Proteins were precipitated with anti-SPL, probed with an antibody specific for pSer94 SPL and then stripped and reprobed with anti-SPL. The graph summarizes 3 experiments (mean ± SEM). (B) Identical to panel A except that the platelets were incubated with 15 µM PGI2 ± okadaic acid.
SPL becomes phosphorylated on serine 94 in platelets incubated with forskolin or PGI2. (A) Human platelets were incubated with 20 µM forskolin (Forsk) ± 1 µM okadaic acid (OA) as indicated. Proteins were precipitated with anti-SPL, probed with an antibody specific for pSer94 SPL and then stripped and reprobed with anti-SPL. The graph summarizes 3 experiments (mean ± SEM). (B) Identical to panel A except that the platelets were incubated with 15 µM PGI2 ± okadaic acid.
To establish whether Ser94 phosphorylation is necessary for PGI2- and forskolin-induced dissociation of the SPL/RGS18/SHP-1 complex, CHO cells were transfected with RGS18 (which they do not normally express) plus either wild-type (WT) SPL or SPL in which serine 94 was replaced with alanine (S94A). SPL was expressed as a Myc-tagged protein. RGS18 was HA-tagged. Lysates from the transfected CHO cells were immunoprecipitated with anti-Myc and immunoblotted with anti-HA. WT SPL coprecipitated with RGS18. Preincubating the CHO cells with either forskolin and okadaic acid or forskolin alone resulted in a 60% decrease in coprecipitation, but had no effect when the cells were transfected with the nonphosphorylatable S94A variant (Figure 4A; supplemental Figure 4). Substituting a phosphomimetic aspartate residue for serine 94 (S94D) caused a reduction in SPL/RGS18 coprecipitation to the same extent as exposing cells express WT SPL to forskolin (Figure 4A; supplemental Figure 3). Identical results were obtained when dissociation of SHP-1 from SPL was measured (Figure 4B). Taken together, these data show that phosphorylation of Ser94 is both necessary and sufficient to account for dissociation of the SPL/RGS18/SHP-1 complex by forskolin.

SPL Ser94 variants affect complex formation and dissociation. (A) CHO cells were transfected with Myc-tagged WT or variant SPL plus HA-tagged RGS18. After 48 hours, the CHO cells were incubated with 20 µM forskolin and 1 µM okadaic acid as indicated. Proteins were precipitated with anti-Myc or nonimmune immunoglobulin (Ig) and then probed with anti-HA before reprobing with anti-Myc. Note that a marker lane was excised as indicated by the vertical line. The graph summarizes 3 experiments (mean ± SEM). The data are expressed relative to normal (WT) SPL. P values are relative to WT SPL. (B) Identical to panel A except the cells were transfected with SHP-1 instead of HA-RGS18 and probed with anti-SHP-1 before reprobing with anti-Myc. NS, nonsignificant.
SPL Ser94 variants affect complex formation and dissociation. (A) CHO cells were transfected with Myc-tagged WT or variant SPL plus HA-tagged RGS18. After 48 hours, the CHO cells were incubated with 20 µM forskolin and 1 µM okadaic acid as indicated. Proteins were precipitated with anti-Myc or nonimmune immunoglobulin (Ig) and then probed with anti-HA before reprobing with anti-Myc. Note that a marker lane was excised as indicated by the vertical line. The graph summarizes 3 experiments (mean ± SEM). The data are expressed relative to normal (WT) SPL. P values are relative to WT SPL. (B) Identical to panel A except the cells were transfected with SHP-1 instead of HA-RGS18 and probed with anti-SHP-1 before reprobing with anti-Myc. NS, nonsignificant.
Inhibition of signaling in transfected CHO cells when RGS18 binding to SPL is blocked
Identification of SPL S94D as a variant that has diminished ability to bind RGS18 provided an opportunity to test the hypothesis that dissociation of the SPL/RGS/SHP-1 complex dampens signaling in agonist- and forskolin/PGI2-stimulated platelets. In the experiments shown in Figure 5, CHO cells were transfected with RGS18 plus either WT or S94D SPL and then stimulated with thrombin. Note that CHO cells normally express RGS10, but not RGS18, so the comparison being made here is on the impact of RGS18 when coexpressed with SPL in forms that either can or cannot bind RGS18. Changes in the cytosolic Ca++ concentration were recorded. Western blots detected equal expression of both forms of SPL. However, the increase in Ca++ was twice as great in the cells expressing WT SPL as S94D SPL.

Overexpressing the SPL S94D variant in CHO cells results in a loss of function. (A) CHO cells were transfected with HA-tagged RGS18 and either Myc-tagged normal (WT) SPL or the phosphomimetic SPL S94D variant. After loading with Fura-2, the cells were stimulated with thrombin and changes in the cytosolic Ca2+ concentration were recorded. (B) Summary of 3 experiments (mean ± SEM). (C) Western blot showing equal expression of WT and S94D.
Overexpressing the SPL S94D variant in CHO cells results in a loss of function. (A) CHO cells were transfected with HA-tagged RGS18 and either Myc-tagged normal (WT) SPL or the phosphomimetic SPL S94D variant. After loading with Fura-2, the cells were stimulated with thrombin and changes in the cytosolic Ca2+ concentration were recorded. (B) Summary of 3 experiments (mean ± SEM). (C) Western blot showing equal expression of WT and S94D.
Inhibition of responses to a thrombin receptor agonist in platelets that express S94D SPL
The results in the CHO cells support the idea that WT SPL, but not S94D SPL, can form complexes with RGS18 that prevent the RGS protein from inhibiting Gq-dependent signaling downstream of thrombin receptors. To determine whether the predicted loss of function occurs when SPL has a reduced ability to sequester RGS18 in platelets, we turned to SPL−/− mice, reconstituting SPL expression in their hematopoietic cells by retroviral transduction. Fetal liver hematopoietic cells were obtained from SPL−/− embryos and transduced with either WT or S94D SPL fused to GFP (Figure 6A). The transduced cells were then transplanted into lethally irradiated WT mice. Expression levels of SPL-GFP and SPL(S94D)-GFP were similar by western blot and flow cytometry (Figure 6B-C). However, platelets expressing the S94D variant showed a reduced aggregation response to the PAR4 (thrombin receptor) agonist peptide, AYPGKF, manifest as a rightward shift in the AYPGKF dose/response curve (Figure 6D). Note that separate experiments established that merging GFP with SPL does not interfere with the binding of RGS18 to SPL, and the S94D variant impairs the binding of RGS18 to SPL-GFP, just as it does to SPL (supplemental Figure 5). Thus, dissociation of the SPL/RGS18/SHP-1 complex and a reduction of the binding of RGS18 to SPL caused by cAMP-dependent phosphorylation results in an increase in free RGS18 and a reduction in G-protein–dependent signaling.

Reconstituting SPL−/− mice with the SPL S94D variant results a loss of function in platelet aggregation. (A) Schematic of the constructs used to express SPL(WT)-GFP, SPL(S94D)-GFP, or GFP alone. LTR, long terminal repeat; pA, polyadenylation signal. (B) Immunoblot of SPL (top) and GFP (bottom) in platelet lysate derived either from SPL−/− mice or from lethally irradiated mice reconstituted with SPL−/− fetal liver cells expressing SPL(WT)-GFP, SPL(S94D)-GFP, or GFP alone. The experiment is representative of 2 similar experiments. (C) Histogram of SPL(WT)-GFP and SPL(S94D)-GFP expression in platelets. The experiment is representative 2 similar experiments. (D) Platelet aggregation stimulated with PAR4 agonist peptide, AYPGKF. Three experiments are summarized in the graph.
Reconstituting SPL−/− mice with the SPL S94D variant results a loss of function in platelet aggregation. (A) Schematic of the constructs used to express SPL(WT)-GFP, SPL(S94D)-GFP, or GFP alone. LTR, long terminal repeat; pA, polyadenylation signal. (B) Immunoblot of SPL (top) and GFP (bottom) in platelet lysate derived either from SPL−/− mice or from lethally irradiated mice reconstituted with SPL−/− fetal liver cells expressing SPL(WT)-GFP, SPL(S94D)-GFP, or GFP alone. The experiment is representative of 2 similar experiments. (C) Histogram of SPL(WT)-GFP and SPL(S94D)-GFP expression in platelets. The experiment is representative 2 similar experiments. (D) Platelet aggregation stimulated with PAR4 agonist peptide, AYPGKF. Three experiments are summarized in the graph.
Discussion
Several decades of research have established the major pathways in the platelet signaling network, but uncertainties remain about how interrelationships between these pathways shape the hemostatic response while avoiding inappropriate platelet activation. Studies that we and others have performed using real-time imaging in the microcirculation suggest that a characteristic architecture emerges during the hemostatic response to injury in which there are regions of greater and lesser platelet activation.31,32 These differences are accompanied by differences in platelet packing density and stability, and are shaped in part by differences in agonist concentration gradients radiating outwards from the actual site of injury.33 That analysis does not fully take into account endogenous limiters of platelet activation, such as RGS proteins, nor the impact of exogenous inhibitors such as PGI2. Yet, these and other regulators of platelet activation affect the hemostatic response as well.34
The studies presented here, along with those of Gegenbauer et al26,27 and Delesque-Touchard et al25 build a picture in which the activity of the platelet signaling network is modulated at an early step that is common to most platelet agonists: that is, the duration of G-protein signaling. Human and mouse platelets express multiple members of the RGS family, 2 of which, RGS10 and RGS18, appear to be the most abundant. Knocking out RGS18 in mice produces a gain of function in platelets,25 as does replacement of normal Gi2α with a variant that is resistant to RGS proteins.24 The phenotype of RGS18 knockout mice suggests that RGS18 and RGS10 are not mutually redundant in platelets, but this conclusion awaits the results of studies on platelets lacking RGS10.
The present studies began with questions about the role of RGS proteins as limiters of platelet activation pathways and evolved to include questions about crosstalk between cAMP-dependent pathways and agonist pathways when platelets encounter PGI2 released from activated or damaged endothelial cells. cAMP is a powerful inhibitor of platelet activation. Drugs that increase platelet cAMP levels, such as dipyridamole, have shown clinical benefit as platelet inhibitors.2,3 Conversely, disrupting the gene that encodes PGI2 receptors in mice produces a prothrombotic phenotype.4 PKA activation by cAMP leads to phosphorylation of a number of platelet proteins35,36 including, as we show here, SPL. As noted in the Introduction, at least 2 scaffold proteins capable of sequestering RGS proteins in platelets have been described. Our own work has focused on SPL and the formation of the SPL/RGS/SHP-1 regulatory complex.8 Spinophilin is able to bind both RGS18 and RGS10, although not both at the same time.8 Spinophilin binds RGS proteins and SHP-1 in resting platelets, releasing them when platelets are activated by thrombin or TxA2, but not by collagen or ADP (Figure 7). Dissociation of SHP-1 makes room for the serine/threonine phosphatase, protein phosphatase 1 (PP1), which binds to SPL in activated platelets.37 Gegenbauer et al26,27 have shown that RGS18 can also bind to a second scaffold protein, 14-3-3γ, in an interaction that is dependent on phosphorylation of RGS18 Ser49 and Ser218. Ser49 phosphorylation increases when platelets are activated and so does the binding of RGS18 to 14-3-3γ. Conversely, phosphorylation of RGS18 decreases, as does binding to 14-3-3γ, when RGS18 undergoes PGI2-stimulated, cAMP-dependent phosphorylation on Ser216.27 Gegenbauer et al suggest that RGS18 is dephosphorylated by PP1. Studies in cells other than platelets show that SPL is a PP1 binding partner.38 We have shown that that PP1 and SHP-1 compete for binding sites on SPL in platelets. As a result, there is little PP1 bound to SPL in resting platelets, but binding of the PP1 α and γ isoforms increases when platelets are activated and SHP-1 dissociates from SPL.37 Forskolin does not cause dissociation of PP1 from SPL (data not shown).

A model for modulating platelet reactivity through regulated control of RGS proteins. In resting platelets, RGS18 is part of a complex that includes SPL and SHP-1 in which SPL is phosphorylated on tyrosines 398 and 483. Addition of thrombin or a TxA2 mimetic causes dissociation of the complex and dephosphorylation of SPL, freeing RGS18. Some of the SPL binds to PP1 once the SPL/RGS18/SHP-1 complex has dissociated (lower left). Addition of PGI2 or forskolin leads to cAMP-dependent phosphorylation of serine 94 in SPL, followed by dissociation of the SPL/RGS18/SHP-1 complex without dephosphorylation of SPL tyrosine residues (lower right). Both events cause an increase in free RGS18 available to bind to activated Gq and Gi, helping to limit platelet activation. Rising cAMP levels also result in phosphorylation of RGS18 Ser216, which Gegenbauer et al27 have shown to displace RGS18 from its binding site on 14-3-3γ (lower middle). AC, adenylyl cyclase; ATP, adenosine triphosphate; DAG, diacylglycerol; PIP, phosphatidylinositol 4,5-bisphosphate; PLC, phospholipase C; SFK, Src family kinases.
A model for modulating platelet reactivity through regulated control of RGS proteins. In resting platelets, RGS18 is part of a complex that includes SPL and SHP-1 in which SPL is phosphorylated on tyrosines 398 and 483. Addition of thrombin or a TxA2 mimetic causes dissociation of the complex and dephosphorylation of SPL, freeing RGS18. Some of the SPL binds to PP1 once the SPL/RGS18/SHP-1 complex has dissociated (lower left). Addition of PGI2 or forskolin leads to cAMP-dependent phosphorylation of serine 94 in SPL, followed by dissociation of the SPL/RGS18/SHP-1 complex without dephosphorylation of SPL tyrosine residues (lower right). Both events cause an increase in free RGS18 available to bind to activated Gq and Gi, helping to limit platelet activation. Rising cAMP levels also result in phosphorylation of RGS18 Ser216, which Gegenbauer et al27 have shown to displace RGS18 from its binding site on 14-3-3γ (lower middle). AC, adenylyl cyclase; ATP, adenosine triphosphate; DAG, diacylglycerol; PIP, phosphatidylinositol 4,5-bisphosphate; PLC, phospholipase C; SFK, Src family kinases.
Combining our observations with those of Gegenbauer and colleagues26,27 suggests that there are 3 states that have to be considered with respect to the availability of RGS18 to interact with activated G proteins and inhibit signaling in platelets (Table 1). We show here that free RGS18 is detectable in resting platelets. We also show that free RGS18 increases when platelets are activated by thrombin and, even more so, when platelets are incubated with PGI2. “Free” is, of course, a relative term, defined functionally here by the ability of GST-Gi2α to capture RGS18 from platelet lysates in the presence of GDP and AlF4−. In resting platelets (state 1), some of the RGS18 is bound to tyrosine-phosphorylated SPL in a heterotrimeric complex that includes SHP-1. Some of the remaining RGS18 is bound to 14-3-3γ, in a complex dependent on serine phosphorylation of RGS18. When platelets are activated by thrombin or TxA2 (state 2), SHP-1 is activated, Y398 and Y483 in SPL become dephosphorylated, and RGS18 dissociates. At approximately the same time, RGS18 serine 49 and 218 phosphorylation increases and the amount of RGS18 bound to 14-3-3γ increases.26 In a sense, SPL and 14-3-3γ are working against each other in activated platelets, one releasing and one capturing RGS18, but the net result is still a net twofold increase in free RGS18, despite the fact that there are more copies of 14-3-3γ than SPL in human and mouse platelets.10,11 State 3 occurs when resting platelets are exposed to PGI2 or forskolin, causing cAMP synthesis to increase. In the presence of cAMP, RGS18 becomes phosphorylated on Ser216 and dissociates from 14-3-3γ.27 At the same time, data presented here show that SPL becomes phosphorylated on Ser94, displacing RGS18, presumably by an allosteric mechanism because the binding site that we have previously mapped for RGS18 does not include Ser94.8 The release of the RGS18 from both of its binding partners contributes to the increase in free RGS18 that we observed in PGI2- and forskolin-stimulated platelets and would be expected to contribute to the well-described resistance of these platelets to activation by platelet agonists. How (if at all) the phosphorylation Ser94 affects the interaction of SPL with other known binding partners that are expressed in platelets, including actin, and whether those interactions affect the sequestration, localization, and release of RGS18, remains to be determined.
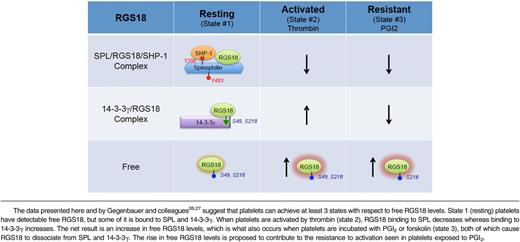
This 3-state model helps to explain how platelet signaling can be dampened, but continue in activated platelets under conditions of vascular injury: RGS18 dissociates from SPL, but some of it is picked up by 14-3-3γ (Table 1). The presence of 2 RGS18 binding partners also helps to account for the extent of loss-of-function phenotype we observed in our studies on SPL−/− mice.8 Platelets from these mice show reduced function in vitro and in vivo, consistent with the idea that there is an increase in free RGS18 levels in the absence of SPL; but the defect is not profound, possibly because of the continued presence of 14-3-3γ. Having said that, it is worth noting that the magnitude of the effect observed in the platelet aggregation assays shown in Figure 6D is not dissimilar from what we have observed in studies on mice lacking SPL or expressing an RGS-insensitive Gi2α mutant,24 and others have observed as a gain of function in mice lacking RGS18.25
Finally, although we have focused here on RGS18, what might be said about RGS10, the other RGS protein that is prominently expressed in platelets? Quantitative proteomics studies suggest that there is at least as much RGS10 as RGS18 in human and mouse platelets.10,11 In resting platelets, RGS10, like RGS18, is bound to SPL, from which it is released when platelets are activated by thrombin and TxA2.8 There is no information available on the binding of RGS10 to 14-3-3γ or any other protein in platelets. The PKA–dependent, phosphorylation-mediated inactivation of RGS10 via nuclear relocation that has been reported in other cell types is presumably not operative in anucleate platelets.39 The phenotype of RGS18 knockout mice suggests that RGS10 and RGS18 are not fully redundant. It remains to be seen whether the knockout of RGS10, either alone or in combination with RGS18, will prove to have an even greater phenotype. It also remains to be established whether RGS10 in platelets is regulated in as complex a manner as RGS18.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by American Heart Association 14SDG20380473 (P.M.) and National Institutes of Health, National Heart, Lung, and Blood Institute R01-HL93123 and P01-HL40387 (L.F.B.). A.J.S. was supported in part by an American Society of Hematology trainee research award.
Authorship
Contribution: P.M., K.O., A.J.S., and H.J. performed experiments; P.M., A.J.S., D.P.S., and L.F.B. designed experiments; and P.M. and L.F.B. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Peisong Ma, Department of Medicine and Pharmacology, University of Pennsylvania, 821 BRB II/III, 421 Curie Blvd, Philadelphia, PA 19104; e-mail: peisong@mail.med.upenn.edu; and Lawrence F. Brass, Department of Medicine and Pharmacology, University of Pennsylvania, 815 BRB II/III, 421 Curie Blvd, Philadelphia, PA 19104; e-mail: brass@mail.med.upenn.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal