

Key Points
In the clinic, all oral antiplatelet medicines have a risk of bleeding complications.
We present an antidote for ticagrelor that reverses its antiplatelet effect in human platelet-rich plasma and its bleeding effect in mice.
Abstract
Ticagrelor is a direct-acting reversibly binding P2Y12 antagonist and is widely used as an antiplatelet therapy for the prevention of cardiovascular events in acute coronary syndrome patients. However, antiplatelet therapy can be associated with an increased risk of bleeding. Here, we present data on the identification and the in vitro and in vivo pharmacology of an antigen-binding fragment (Fab) antidote for ticagrelor. The Fab has a 20 pM affinity for ticagrelor, which is 100 times stronger than ticagrelor’s affinity for its target, P2Y12. Despite ticagrelor’s structural similarities to adenosine, the Fab is highly specific and does not bind to adenosine, adenosine triphosphate, adenosine 5′-diphosphate, or structurally related drugs. The antidote concentration-dependently neutralized the free fraction of ticagrelor and reversed its antiplatelet activity both in vitro in human platelet-rich plasma and in vivo in mice. Lastly, the antidote proved effective in normalizing ticagrelor-dependent bleeding in a mouse model of acute surgery. This specific antidote for ticagrelor may prove valuable as an agent for patients who require emergency procedures.
Introduction
Acute coronary syndrome (ACS) is one of the most common causes of cardiovascular mortality and morbidity worldwide, and dual-antiplatelet therapy, consisting of aspirin and a P2Y12 antagonist, is critical for the treatment of ACS patients. Among the P2Y12 antagonists, ticagrelor is unique because it is direct acting and binds reversibly. In contrast, the thienopyridines (ticlopidine, clopidogrel, and prasugrel) are all prodrugs and bind irreversibly to P2Y12.1 In the phase III clinical trial Platelet Inhibition and Patient Outcomes, ticagrelor, when compared to clopidogrel, decreased the incidence of major adverse cardiovascular events and total mortality in patients with ACS when given in addition to aspirin.2 Ticagrelor is now preferred over clopidogrel for the management of non-ST-elevation ACS patients who undergo an early invasive or ischemia-guided strategy or those who receive a coronary stent.3 In addition, ticagrelor, like prasugrel, has a class Ib recommendation for patients with ST-segment elevation and, therefore, should be considered an option for patients with any ACS.4
Despite the improvements in treatment with ticagrelor, as for all antiplatelet medicines, there is an increased risk of bleeding complications. In the Platelet Inhibition and Patient Outcomes trial, 11.6% of ticagrelor patients and 11.2% of clopidogrel patients suffered major bleeding, of which 5.8% for both groups was classified as fatal or life-threatening. In addition, with ticagrelor, there was a higher rate of major bleeding not related to coronary artery bypass grafting.2 Currently, there are limited treatment options for these patients, including platelet transfusions and supportive care. Although platelet transfusions restore platelet function in patients on aspirin,5 they have shown no or minimal ability to reverse adenosine 5′-diphosphate (ADP)-induced aggregation in healthy volunteers treated with clopidogrel6 or in ticagrelor-treated patients.7,8
Because both antiplatelet and anticoagulant therapies are known to increase the risk of bleeding complications, specific antidotes are desired in the clinical situations of life-threatening bleeding or urgent surgery where patients cannot wait for the drugs’ effects to stop naturally.9 Recently, specific antidotes for anticoagulants have been described and are undergoing clinical trials. These antidotes include a specific neutralizing antigen-binding fragment (Fab) designed to reverse the thrombin inhibitor dabigatran,10 a recombinant catalytically inactive factor Xa (FXa) that should reverse any of the FXa inhibitors,11 and a small molecule that can reverse the FXa inhibitors and dabigatran.12 In this study, we report an antidote for ticagrelor, designed (1) to be highly specific and to neutralize both ticagrelor and ticagrelor’s active metabolite (TAM), which has similar P2Y12 potency and is present at 30% to 40% of ticagrelor levels in the circulation of patients13 ; and (2) to have a half-life similar to that of ticagrelor and TAM in humans, which is 9.8 and 12.4 hours, respectively.14 A human Fab was selected as the most appropriate format for the antidote because this would confer the appropriate specificity and be expected to have a half-life of ∼12 hours in patients. Herein we describe the engineering and x-ray crystal structure of the Fab MEDI2452 that binds and neutralizes ticagrelor and TAM, and that reverses both the antiplatelet activity and the drug-induced bleeding in mouse models.
Materials and methods
Antibody isolation and engineering
To enable antibody isolation, screening, and engineering, 6 different haptens were synthesized. These included unmodified ticagrelor, TAM, and ticagrelor inactive metabolite (TIM) (Figure 1A). TIM is the main ticagrelor metabolite in the urine but has minimal exposure in the circulation15 and has no significant activity on P2Y12 (data not shown). Derivatives of ticagrelor, TIM, and adenosine were conjugated to biotin via a short triamide linker to create biotinylated ticagrelor, TIM, and adenosine. Antibodies were isolated from a human single-chain variable-fragment phage display library. Detailed information on high-throughput expression and specificity screening is compiled in supplemental Methods, available on the Blood Web site. A representative number of individual clones were expressed as Fabs and characterized for specificity. To engineer antibody affinity, large single-chain variable fragment libraries derived from clone 72 were created by oligonucleotide-directed mutagenesis of the variable heavy (VH) or variable light (VL) chain complementarity-determining region (CDR)3. The libraries were subjected to affinity-based phage display selections to select variants with a higher affinity to ticagrelor and TAM. Detailed information on affinity screening is compiled in supplemental Methods. The highest-affinity antibodies were sequenced, and a representative number of individual clones were expressed as Fabs.

Specificity of Fabs assessed by competitive inhibition of Fab binding to biotinylated ticagrelor. (A) Structures of ticagrelor, TAM, TIM, and biotinylated ticagrelor. The substituent R groups of ticagrelor are indicated. (B) Specificity profile of Fab 72. (C) Specificity profile of MEDI2452. Data shown are for ticagrelor, TAM, TIM, adenosine, ADP, ATP, and 3 of the 12 structurally related compounds tested for clarity. No binding was detected to any of the 12 structurally related compounds at concentrations up to 0.1 mM. Data are mean ± standard error of the mean (SEM) for 3 replicates.
Specificity of Fabs assessed by competitive inhibition of Fab binding to biotinylated ticagrelor. (A) Structures of ticagrelor, TAM, TIM, and biotinylated ticagrelor. The substituent R groups of ticagrelor are indicated. (B) Specificity profile of Fab 72. (C) Specificity profile of MEDI2452. Data shown are for ticagrelor, TAM, TIM, adenosine, ADP, ATP, and 3 of the 12 structurally related compounds tested for clarity. No binding was detected to any of the 12 structurally related compounds at concentrations up to 0.1 mM. Data are mean ± standard error of the mean (SEM) for 3 replicates.
Production of Fabs
Separate heavy- and light-chain expression plasmids were used for transient transfection of a CHOK1 cell line adapted for suspension culture.16 After 7 days, the cells were harvested by centrifugation and the supernatants were filtered and purified using CaptureSelect IgG-CH1 (Life Technologies, Carlsbad, CA), following the manufacturer’s recommendations.
Specificity assays
To identify compounds that have some similarity to ticagrelor, in addition to adenosine, ADP, and adenosine triphosphate (ATP), a structural database for marketed drugs17 was interrogated. Methods of searching included 2-dimensional fingerprint similarity and 3-dimensional shape and electrostatic similarity based on ticagrelor x-ray crystallography and nuclear magnetic resonance conformations. Specific binding of purified antibodies to ticagrelor and other compounds was analyzed in a homogeneous time-resolved fluorescence competition assay in which unlabeled compounds compete for Fab binding to biotinylated ticagrelor. When compounds were dissolved in dimethylsulfoxide (DMSO), data were normalized to correct for any such vehicle-related effects using parallel titrations of DMSO alone.
Affinity measurements
The affinities of Fabs were determined with the Octet RED384 System (ForteBio, Menlo Park, CA) or the KinExA 3200 Instrument (Sapidyne Instruments, Boise, ID). A constant concentration of Fab was incubated with a dilution series of ticagrelor and allowed to equilibrate for 3 to 5 days. The concentration of free Fab was determined by capturing unbound Fab with biotinylated ticagrelor on streptavidin biosensors (using the Octet system) or with biotinylated ticagrelor on streptavidin beads (using the KinExA instrument). The equilibrium KD for the antiticagrelor Fab was determined using the constant partner analysis.
Protein crystallography
Detailed information on the complex formation, crystallization, and crystallography is compiled in supplemental Methods.
Human platelet aggregation
Citrate-anticoagulated blood was collected from healthy donors. Platelet-rich plasma (PRP) was preincubated with 1 μM ticagrelor, 1 μM TAM, or vehicle (5% mannitol or DMSO, respectively) for 1 hour before co-incubation with MEDI2452 or vehicle (phosphate-buffered saline) for 30 minutes, followed by initiation of aggregation by 20 μM ADP. Light transmission aggregometry was evaluated using the PAP-8 (Bio/Data, Horsham, PA) aggregometer, and data for final aggregation (FA) at 6 minutes was recorded and used for calculations of both MEDI2452 half-maximum reversal (IC50) and percentage reversal: 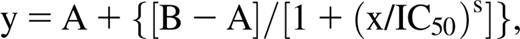 where y is FA, A is minimum FA, B is maximum FA, s is the slope of the concentration response curve, and x is MEDI2452 concentration. Percentage reversal was calculated as:
where y is FA, A is minimum FA, B is maximum FA, s is the slope of the concentration response curve, and x is MEDI2452 concentration. Percentage reversal was calculated as:
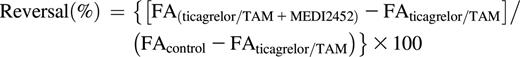
Subsamples of PRP collected after 30 minutes of co-incubation with MEDI2452 but before ADP-induced aggregation was used for analysis of free-ticagrelor plasma concentration.
Mouse platelet aggregation in ticagrelor-treated mice
Mice were pretreated with a bolus dose of ticagrelor (1200 μg/kg) or vehicle (5% mannitol) given over 5 minutes, followed by a continuous infusion (30 μg/kg/min) for 15 minutes. After stop of infusion (time [t] = 0), a bolus dose of MEDI2452 (250 mg/kg), vehicle (phosphate-buffered saline), or antibody isotype control was given over 45 seconds. Terminal blood samples for platelet aggregation were collected at t = 0, 5, 15, 30, and 60 minutes from the abdominal aorta. Antibody isotype control was tested only at the 30-minute time point to demonstrate that it had no effect and to limit the number of mice in the study. Samples for total plasma exposure were also collected at t = 0. Whole blood impedance aggregometry was evaluated using the Multiplate impedance aggregometer (Dynabyte, Munich, Germany), and data for the mean area under the curve (AUC) of aggregation units measured over time were recorded. Extent of reversal was calculated from the AUC data as described for the light transmission aggregometry FA data.
Mouse bleeding in ticagrelor-treated mice
Mice were pretreated with a continuous infusion of ticagrelor (300 μg/kg/min) or vehicle for 20 minutes. After stop of infusion (t = 0), a bolus dose of MEDI2452 (600 mg/kg) or vehicle (histidine sucrose buffer) was given over 45 seconds. At t = 30 minutes, bleeding was induced by cutting 5 mm from the tip of the tail. The tip of the tail was rinsed with water (2 mL/min), and the blood and water mixture was collected in a small chamber, where a stirrer mixed the fluid to enhance hemolysis and establish a homogeneous solution. Light transmission, at 525 nm, was recorded for 30 minutes when terminal blood samples were collected from the abdominal aorta for platelet aggregation. Light transmittance was transformed to absorbance and used to calculate blood loss as area under the absorbance curve and total bleeding time by plotting absorbance over time. All transmittance below 95% was defined as bleeding. Samples for platelet aggregation and total and free plasma exposure were also collected at end of ticagrelor infusion (t = 0) and at time of tail cut (t = 30 minutes). Statistical differences between treatment groups were evaluated using the Mann-Whitney nonparametric analysis.
Total- and free-ticagrelor plasma concentrations
Total plasma concentration of ticagrelor and TAM and the concentration of free unbound ticagrelor in plasma were measured as described in supplemental Methods.
Results
Antibody isolation, specificity screening, and engineering
Antibodies were isolated from a human antibody phage library by selection on biotinylated ticagrelor, followed by screening for specificity. The chemical starting point for the development of ticagrelor was ATP, and it retains an adenosine-like core,18 so initial specificity screens included direct binding to biotinylated ticagrelor, biotinylated adenosine, and unmodified ticagrelor in a competition assay (supplemental Figure 1). A panel of 80 antibodies that bound ticagrelor but not adenosine was taken forward for further profiling. A structural database for marketed drugs was also interrogated for molecules that have any structural similarity to ticagrelor. From this in silico analysis, a panel of 12 compounds was selected that importantly included 6 potential co-medications. The structures of these compounds are shown in supplemental Figure 2. The ability of these compounds to competitively inhibit the binding of antibody to biotinylated ticagrelor was tested. From this, we identified Fab 72, which showed no binding to adenosine or the 12 structurally related compounds (Figure 1B). Fab 72 functionally neutralized ticagrelor’s and TAM’s inhibition of ADP-induced P2Y12 signaling (supplemental Figure 3) and had binding affinity of approximately 7 nM for both compounds (Table 1).
Affinity and CDR3 sequences of the parental Fab 72 and optimized variants
| Antibody | VH CDR3 | VL CDR3 | Hapten | Equilibrium, KD | 95% Confidence interval |
|---|---|---|---|---|---|
| Fab 72 | GSHLY99DFW100bSASHPPNDALAI | GTW91D92I93S94LSAGL | Ticagrelor | 7.4 nM | 1.8-21.5 nM |
| Fab 152 | GSHLYDFWSASHPPNDALAI | GTWLYDRAVGL | Ticagrelor | 43.2 pM | 2.8-119.2 pM |
| Fab 162 | GSFDYYFWSASHPPNDALAI | GTWDISLSAGL | Ticagrelor | 162.5 pM | 125.4-206.3 pM |
| MEDI2452 | GSFDYYFWSASHPPNDALAI | GTWLYDRAVGL | Ticagrelor | 19.6 pM | 13.0-28.7 pM |
| TAM | 19.7 pM | 4.9-44.7 pM | |||
| TIM | ∼20 nM |
| Antibody | VH CDR3 | VL CDR3 | Hapten | Equilibrium, KD | 95% Confidence interval |
|---|---|---|---|---|---|
| Fab 72 | GSHLY99DFW100bSASHPPNDALAI | GTW91D92I93S94LSAGL | Ticagrelor | 7.4 nM | 1.8-21.5 nM |
| Fab 152 | GSHLYDFWSASHPPNDALAI | GTWLYDRAVGL | Ticagrelor | 43.2 pM | 2.8-119.2 pM |
| Fab 162 | GSFDYYFWSASHPPNDALAI | GTWDISLSAGL | Ticagrelor | 162.5 pM | 125.4-206.3 pM |
| MEDI2452 | GSFDYYFWSASHPPNDALAI | GTWLYDRAVGL | Ticagrelor | 19.6 pM | 13.0-28.7 pM |
| TAM | 19.7 pM | 4.9-44.7 pM | |||
| TIM | ∼20 nM |
Changes in sequences from Fab 72 are in bold type. Key residues relating to Figure 2 are followed by Kabat numbers in superscript.
To increase the affinity of Fab 72, phage display libraries were generated by randomizing amino acids in the VH and VL CDR3 loops, followed by affinity selection. The highest-affinity VH and VL CDR3 variants, Fab 162 and Fab 152, respectively, were combined to create the anti-ticagrelor/TAM Fab, MEDI2452. The KD of MEDI2452 binding to ticagrelor is 20 pM, which was 350-fold improved over the parent Fab 72 (Table 1). The specificity of MEDI2452 was explored using competitive inhibition as described earlier. The Fab demonstrated equivalent binding to ticagrelor and TAM and weak binding to TIM (Figure 1C). In addition, MEDI2452 demonstrated no binding to adenosine, ADP, ATP, or any 12 structurally related drugs, indicating that it is ticagrelor and TAM specific. The lack of binding to adenosine was confirmed in a direct binding assay.
Ticagrelor Fab structure
The structure of Fab 72 in complex with ticagrelor was determined to 1.7-Å resolution (Figure 2A). The CDRs form a highly concave surface, and ticagrelor is inserted deep into the interface between VH and VL domains. This type of binding is commonly observed for small haptens.19 All CDRs except VL CDR2 are contributing directly to ticagrelor binding, and a large portion of VH CDR3 is disordered. Ticagrelor’s difluorophenyl group is located in a cavity lined with hydrophobic residues, including vernier residues VH Trp47, VL Phe98, and VH CDR3 residue Leu100L. A key residue in the interaction with ticagrelor is VL Trp91, which is involved in both a π-stacking against the adenosine-like core and a hydrogen bond to one of the ribose hydroxyl groups at the cyclopentyl moeity. Additional interactions to the adenosine-like core are provided by VH CDR1 His35 and VH CDR3 Tyr99. The thiopropyl substituent stacks against the main chain of the VH CDR2 loop. The hydroxyethyl substituent on the cyclopentyl moiety protrudes into the solvent and does not make any interactions with the Fab.

Crystal structures of Fab 72 and MEDI2452 in complex with ticagrelor. Fab 72 (pdb id 5alc) (A) and MEDI2452 (pdb id 5alb) (B) are shown in ribbon representation, with amino acid residues within 7 Å from ticagrelor shown as sticks. Some main-chain atoms were omitted for clarity. Light chains are shown in beige; heavy chains in light blue; CDR3s from both chains in green. VH CDR3 could not be modeled in the Fab 72 structure, and a tentative location is drawn as a dashed line. The orange arrow indicates the shift in VL CDR3 observed in MEDI2452 compared to Fab 72. Residues are numbered following the Kabat scheme and are prefixed with L or H to indicate light or heavy chain, respectively.
Crystal structures of Fab 72 and MEDI2452 in complex with ticagrelor. Fab 72 (pdb id 5alc) (A) and MEDI2452 (pdb id 5alb) (B) are shown in ribbon representation, with amino acid residues within 7 Å from ticagrelor shown as sticks. Some main-chain atoms were omitted for clarity. Light chains are shown in beige; heavy chains in light blue; CDR3s from both chains in green. VH CDR3 could not be modeled in the Fab 72 structure, and a tentative location is drawn as a dashed line. The orange arrow indicates the shift in VL CDR3 observed in MEDI2452 compared to Fab 72. Residues are numbered following the Kabat scheme and are prefixed with L or H to indicate light or heavy chain, respectively.
In the structure of the affinity-improved Fab, MEDI2452, the ticagrelor binding is similar to that of Fab 72, with all the interactions mentioned above retained but with some important differences (Figure 2B). The combination of the VL CDR3 mutations Asp92Leu and Ser94Asp breaks a hydrogen bond within the VL CDR3 loop to create a more “relaxed” structure. The new conformation is correlated with a 15-degree tilt of the pyrimidine ring about the attachment of the cyclopropyl-difluorophenyl substituent. The new position of the pyrimidine ring brings it approximately 0.2 Å closer to VH CDR3 Tyr99 in MEDI2452 compared to Fab 72. Moreover, VL Ile93Tyr introduces a hydrogen bond donor, which makes interactions with the VH CDR3 loop, thus defining the binding site further.
Reversal of ADP-induced human platelet aggregation
In human PRP treated with ticagrelor or TAM, MEDI2452 reversed the antiplatelet effect of both in a concentration-dependent manner (Figure 3A). Its potency (IC50) to reverse 1 μM ticagrelor or 1 μM TAM was 0.64 and 0.78 μM (n = 5), respectively. Maximal reversals of 78% and 62% (n = 5) for ticagrelor and TAM, respectively, were achieved with concentrations of MEDI2452 up to 1 μM as expected, given that MEDI2452 binds 1:1 with ticagrelor and TAM. In parallel, the concentration of free ticagrelor in human PRP was measured. MEDI2452 reduced the free-ticagrelor concentration, with an IC50 value of 0.49 μM (Figure 3B), which inversely correlated with restoration of ADP-induced platelet aggregation.

MEDI2452 reversal of ADP-induced human platelet aggregation in vitro. (A) MEDI2452 concentration-dependent reversal of 1 μM ticagrelor (○) or 1 μM TAM (●) mediated inhibition of 20 μM ADP-induced aggregation. (B) Reduction of free-ticagrelor concentration in plasma in the presence of 1 μM ticagrelor. Mean (n = 5) ± SEM.
MEDI2452 reversal of ADP-induced human platelet aggregation in vitro. (A) MEDI2452 concentration-dependent reversal of 1 μM ticagrelor (○) or 1 μM TAM (●) mediated inhibition of 20 μM ADP-induced aggregation. (B) Reduction of free-ticagrelor concentration in plasma in the presence of 1 μM ticagrelor. Mean (n = 5) ± SEM.
Reversal of ex vivo ADP-induced aggregation in ticagrelor-treated mice
Mice were infused with ticagrelor to achieve respective mean ticagrelor and TAM plasma concentrations of 1.4 and 0.09 μM at stop of infusion (t = 0). This induced a next-to-complete inhibition of ADP-induced aggregation at all time points assessed (5, 30, and 60 minutes) after stop of infusion. The aggregation response in the vehicle- and ticagrelor-treated mice ranged from 432-494 and from 2-15 aggregation units (AU)*min (area under the aggregation curve), respectively (Figure 4A). Immediately after stopping the ticagrelor infusion, MEDI2452 was dosed as a single bolus, and ADP-induced aggregation was measured. MEDI2452 reversed the antiplatelet effect of ticagrelor by 34%, 94%, and 84% at 5, 30, and 60 minutes postadministration, respectively (Figure 4), whereas the isotype control resulted in no reversal at 30 minutes postadministration. Thus, the full onset of reversal required 30 minutes after antidote administration.

MEDI2452 reversal of ADP-induced whole blood aggregation ex vivo. (A) Individual data for each treatment group after the stop of ticagrelor infusion. Vehicle control (▪), ticagrelor alone (●), ticagrelor plus MEDI2452 (○), and ticagrelor plus isotype control (Δ). Open bar represents mean data (n = 4). AU*min, area under aggregation curve per minute. At 15 minutes, data were collected only for the ticagrelor-plus-MEDI2452 group. (B) Percentage reversal induced by MEDI2452; mean data (n = 4) ± SEM.
MEDI2452 reversal of ADP-induced whole blood aggregation ex vivo. (A) Individual data for each treatment group after the stop of ticagrelor infusion. Vehicle control (▪), ticagrelor alone (●), ticagrelor plus MEDI2452 (○), and ticagrelor plus isotype control (Δ). Open bar represents mean data (n = 4). AU*min, area under aggregation curve per minute. At 15 minutes, data were collected only for the ticagrelor-plus-MEDI2452 group. (B) Percentage reversal induced by MEDI2452; mean data (n = 4) ± SEM.
Reduction of bleeding in ticagrelor-treated mice
To translate the effect of MEDI2452 on platelet aggregation to a potential effect on bleeding, a prophylactic mouse tail bleeding study was performed. Ticagrelor was infused to mean total ticagrelor and TAM plasma concentrations of 7.6 and 0.3 μM, respectively, to provide a significant drug-dependent bleeding window. Immediately after stopping the ticagrelor infusion, MEDI2452 was dosed as a single bolus of 600 mg/kg over 30 seconds. After 30 minutes, ADP-induced aggregation was fully normalized and the mean ticagrelor-free plasma concentration was reduced from 4.7 nM to below 0.03 nM (lower limit of quantification). Bleeding was initiated by a tail cut and monitored for 30 minutes. In vehicle-treated mice, the mean total ticagrelor and TAM plasma concentrations at time of tail cut were 2.4 and 0.6 μM, respectively. Total blood loss and bleeding time over 30 minutes were significantly (P < .05) enhanced by ticagrelor: ∼3.8-fold and ∼1.6-fold, respectively. MEDI2452 significantly (P < .05) reversed blood loss and bleeding time relative to ticagrelor alone, and reversed them to levels not significantly different from those seen in mice not treated with ticagrelor (Figure 5).

MEDI2452 reversal of ticagrelor-induced bleeding. Individual data for total blood loss (A) and total bleeding time (B). Vehicle control (▪), ticagrelor alone (●) and ticagrelor plus MEDI2452 (○). Filled bar represents mean data (n = 12).
MEDI2452 reversal of ticagrelor-induced bleeding. Individual data for total blood loss (A) and total bleeding time (B). Vehicle control (▪), ticagrelor alone (●) and ticagrelor plus MEDI2452 (○). Filled bar represents mean data (n = 12).
Discussion
We have designed a specific antidote for ticagrelor using phage display and specificity screening to isolate and engineer a human Fab, MEDI2452. The affinity of MEDI2452 for ticagrelor and TAM is 20 pM, which is 100-fold greater than the 2 nM affinity of ticagrelor for its receptor P2Y12.18 The crystal structure for MEDI2452 shows ticagrelor bound in a deep crevice between the VH and VL interface. The design strategy of labeling ticagrelor with biotin via a triamide linker to the hydroxyethyl group is substantiated because the crystal structure demonstrates that the hydroxyethyl group is not involved in any interaction with MEDI2452. This is further supported by the fact that the Fab also binds TAM (which lacks the hydroxyethyl group) with identical affinity. MEDI2452 shows weak binding to TIM, which lacks the cyclopropyl-difluorophenyl group. In the MEDI2452-ticagrelor complex, the cyclopropyl-difluorophenyl group is buried at the bottom of the hydrophobic pocket and must play a key structural role in aligning the ticagrelor adenosine-like core with the VL CDR3 residue Trp L91. Because the chemical starting point for ticagrelor was ATP and it retains an adenosine-like core, a critical attribute for antidote specificity was to demonstrate no binding of adenosine. The lead isolation strategy involved both high-throughput and detailed specificity analysis, and no binding of adenosine was detected by either competitive or direct-binding analyses. From the structural analysis, the purine ring and ribose group of adenosine might be expected to mimic the interaction of ticagrelor’s adenosine-like core. However, the lack of binding may be explained by the absence of 2 hydrophobic R groups (cyclopropyl-difluorophenyl and thiopropyl), which significantly reduces the relative shape complementarity and the hydrophobicity of the binding interaction. Despite the recent publication of the P2Y12 structure in complex with an antagonist,20 little is known about how ticagrelor binds P2Y12; therefore, we cannot compare the binding of Fab and P2Y12 to ticagrelor.
The high affinity and specificity of MEDI2452 was achieved by CDR-directed protein engineering and affinity screening. An analysis of the structures of the parental Fab 72 and the lead MEDI2452 shows the significance of some of the changes introduced during the affinity maturation. The mutations in VL CDR3 appear to have particularly high impact, resulting in a different loop conformation and an additional hydrogen bond in MEDI2452 to define the binding cavity. In contrast, contributions from mutations in the VH CDR3 are structurally less obvious. These observations from the structure are partly confirmed by data for modified antibodies containing modifications in VL CDR3 (Fab 152) or VH CDR3 (Fab 162) only that resulted in 200-fold and 50-fold improvement over Fab 72, respectively. However, although the VL CDR3 changes appear to have a greater effect, both sets of mutations add significant improvement. It should be noted that the crystal structure is a static picture of the complex and is not able to capture any of the protein and ligand dynamics involved in binding.
In patients, the minimum-to-maximum ticagrelor and TAM plasma concentrations after 4 weeks of ticagrelor treatment (90 mg twice daily) has been documented as 0.4 to 1.5 μM and 0.2 to 0.5 μM, respectively.13 To demonstrate the relative potency and clinical relevance of the antidote, human PRP was preincubated with 1 μM ticagrelor or 1 μM TAM in vitro, followed by co-incubation with MEDI2452 before assessing ADP-induced aggregation. As expected, given a 1:1 binding relationship, as visualized in the cocrystal structure, maximal reversal occurred at and above 1 μM of MEDI2452 for both ticagrelor and TAM. MEDI2452 had similar potency to reverse both ticagrelor and TAM (IC50 values were not significantly different, P = .47), as predicted by the equivalent affinities. Both ticagrelor and TAM have high plasma protein binding (99.8%),21 which explains why the free-ticagrelor concentration under the conditions evaluated in the in vitro experiments was ∼2 nM despite a total concentration of 1 μM. The restoration of ADP-induced platelet aggregation was matched by the elimination of free ticagrelor from the plasma, with <0.05 nM free ticagrelor remaining at and above 1 μM MEDI2452. Thus, these in vitro experiments in human PRP provided proof of concept for the antidote, with the restoration of platelet aggregation, and also proof of mechanism in the elimination of free ticagrelor. Ticagrelor has, in addition to P2Y12 antagonism, been shown to have an additional mechanism, in that it inhibits the adenosine transporter ENT1.22 Because ticagrelor’s affinity is weaker for ENT1 than it is for P2Y12, it can be predicted that the ENT1 inhibition is maximally reversed at lower or similar MEDI2452 exposures that maximally reverse platelet aggregation.
A mouse ex vivo platelet aggregation study was used to translate these in vitro data into the in vivo setting. Ticagrelor was dosed to a total plasma exposure of 1.4 μM, providing complete inhibition of ADP-induced aggregation. TAM was present at about 35% of the levels of ticagrelor (0.07 vs 0.2 μM) at 60 minutes, similar to the relationship in plasma from patients. MEDI2452 (250 mg/kg) effectively and next-to-completely reversed ticagrelor-induced inhibition of ADP-induced aggregation ex vivo at 30 and 60 minutes postdosing but demonstrated only partial reversal (34%) after 5 minutes. The effective onset time of 30 minutes can be rationalized by 2 factors: (1) the off-rate of ticagrelor from P2Y12 is 14 minutes1 and (2) the high plasma protein binding of ticagrelor is >99.8%.21 Because MEDI2452 is unlikely to bind ticagrelor or TAM when they are bound to P2Y12 or plasma protein, the action of MEDI2452 is to bind and neutralize the <0.2% fraction that is free at any given time point. MEDI2452 therefore requires time to act, because the remaining ticagrelor and TAM dissociates from both the plasma proteins and P2Y12 until nearly all ticagrelor and TAM is bound to MEDI2452 and no free fraction remains. The in vitro data in human PRP indicate that maximal reversal of aggregation was achieved after the free fraction was reduced below 0.05 nM and, in mice, complete reversal of ADP aggregation was achieved with a dose of MEDI2452 that eliminated the free-ticagrelor plasma concentration. Because one of the intended indications for MEDI2452 is as an antidote for ticagrelor patients requiring urgent surgery, MEDI2452 was evaluated in a mouse bleeding experiment designed to mimic the clinical setting were complete reversal should be achieved before initiation of surgery. In this prophylactic setting, MEDI2452 rapidly normalized ticagrelor-dependent bleeding. The onset time of 30 minutes, translated from the ex vivo model to this in vivo model, demonstrated that MEDI2452 can normalize both blood loss and bleeding time to those of mice not treated with ticagrelor.
In conclusion, MEDI2452 is a high-affinity specific Fab for ticagrelor and its active metabolite (TAM). In a concentration-dependent manner, it neutralizes the free ticagrelor and TAM, reversing both ticagrelor- and TAM-mediated inhibition of platelet aggregation in human PRP in vitro and in ticagrelor-treated mice in vivo. Lastly, MEDI2452 was shown to normalize bleeding in ticagrelor-treated mice when given before induction of bleeding, simulating an acute surgery situation. The pharmacologic profile of MEDI2452 warrants further preclinical and clinical investigation. If successful, this Fab has the potential to be a valuable agent for ticagrelor patients to treat both the rare emergency of life-threatening bleeding and the need for urgent surgery.
The online version of this article contains a data supplement.
There is an on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Gareth Rees for technical assistance with affinity assessment, Cameron Bowden for help in preparing figures, and the Biopharmaceutical Development teams for Fab expression and purification expertise.
Authorship
Contribution: A.B., T.I., T.S., S.N., M.P., P.G., T.V., and G.H. designed the research; S.P., A.J., A.-S.S., L.O., P.S., P.N., F.K., M.A., and J.S. performed the research; T.A. provided vital new reagents; and A.B., T.I., T.S., and S.N. wrote the manuscript.
Conflict-of- interest disclosure: All authors are employees of AstraZeneca and MedImmune.
The current affiliation for P.S. is Integrative Research Laboratories, Göteborg, Sweden.
Correspondence: Andrew Buchanan, MedImmune R&D, Granta Park, Cambridge CB22 6GH, United Kingdom; e-mail: buchanana@medimmune.com; and Sven Nylander, AstraZeneca R&D Mölndal, Pepparedsleden 1, 43183 Mölndal, Sweden; e-mail: sven.nylander@astrazeneca.com.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal