

Abstract
Mutations in the Bcr-Abl kinase domain may cause, or contribute to, resistance to tyrosine kinase inhibitors (TKIs) in chronic myeloid leukemia patients. Recommendations aimed to rationalize the use of BCR-ABL mutation testing in chronic myeloid leukemia have been compiled by a panel of experts appointed by the European LeukemiaNet (ELN) and European Treatment and Outcome Study and are here reported. Based on a critical review of the literature and, whenever necessary, on panelists' experience, key issues were identified and discussed concerning: (1) when to perform mutation analysis, (2) how to perform it, and (3) how to translate results into clinical practice. In chronic phase patients receiving imatinib first-line, mutation analysis is recommended only in case of failure or suboptimal response according to the ELN criteria. In imatinib-resistant patients receiving an alternative TKI, mutation analysis is recommended in case of hematologic or cytogenetic failure as provisionally defined by the ELN. The recommended methodology is direct sequencing, although it may be preceded by screening with other techniques, such as denaturing-high performance liquid chromatography. In all the cases outlined within this abstract, a positive result is an indication for therapeutic change. Some specific mutations weigh on TKI selection.
Introduction
Soon after the advent of the Bcr-Abl tyrosine kinase inhibitor (TKI) imatinib mesylate for the treatment of chronic myeloid leukemia (CML) in 2001, it became clear that Philadelphia-positive (Ph+) cells could evolve to elude inhibition. Since the first imatinib-resistant cases, point mutations in the kinase domain (KD) of Bcr-Abl were identified1-6 that could impair or even totally abrogate imatinib binding.7-10 Over the last decade, intensive efforts have been spent in the characterization of the biologic and clinical significance of these mutations on one hand and in the development of novel inhibitors retaining efficacy against as many Bcr-Abl mutant forms as possible on the other hand. The list of amino acid substitutions detected in imatinib-resistant patients has steadily grown to include > 90 different ones (Figure 1), although some are definitely more frequent than others. Different mutations have been shown to confer variable degrees of resistance to imatinib.11 Clinical experience with dasatinib and nilotinib, the second-generation TKIs having received market approval so far, has demonstrated that definite, much narrower spectra of mutations retain insensitivity to these agents—and these spectra are nonoverlapping, the T315I being the unique exception.12-17 As more and more therapeutic options to consider for patients who do not achieve an optimal response to imatinib have become accessible, BCR-ABL KD mutation analysis has turned into a more and more useful tool for physicians. Other mechanisms of resistance are known to exist18 and to not necessarily be mutually exclusive with mutations. Notwithstanding, the knowledge of the Bcr-Abl KD mutation status is a valuable piece of information to be integrated in the decision algorithm aimed at tailoring the best therapeutic strategy for each of these patients: increasing imatinib dose,19-22 switching to the second-generation TKIs dasatinib or nilotinib,12-17,23-26 moving to allogeneic stem cell transplantation,27 or testing an investigational compound. Mutation analysis of the BCR-ABL KD is now being performed in a growing number of laboratories. However, there is still considerable confusion among physicians as to when mutation analysis should be prescribed, which techniques provide the most informative results, and how these results should be interpreted and translated into clinical decisions. Although we are aware that the therapeutic scenario is in continuous evolution and that some issues at present remain controversial, we think that sufficient information is available to compile a series of recommendations aimed to optimize the use of this test in the context of routine management of CML patients.

Map of all the amino acid substitutions in the Bcr-Abl KD identified in clinical samples from patients reported to be resistant to imatinib in published papers. Key structural motifs within the KD are indicated. P-loop indicates phosphate binding loop; SH2 contact and SH3 contact, contact regions with SH2 and SH3 domain-containing proteins; and A-loop, activation loop. Star indicates amino acid position reported to be directly involved in imatinib binding via hydrogen bonds or van der Waals interactions.7 K247R and Y320C are in italic because they have been reported to be single nucleotide polymorphisms. Numbering of residues is according to Abl Ia isoform. Data were collated from 27 studies published between 2001 and 2009.1-6,14,16,17,28,35,51-53,61,62,72,82-91
Map of all the amino acid substitutions in the Bcr-Abl KD identified in clinical samples from patients reported to be resistant to imatinib in published papers. Key structural motifs within the KD are indicated. P-loop indicates phosphate binding loop; SH2 contact and SH3 contact, contact regions with SH2 and SH3 domain-containing proteins; and A-loop, activation loop. Star indicates amino acid position reported to be directly involved in imatinib binding via hydrogen bonds or van der Waals interactions.7 K247R and Y320C are in italic because they have been reported to be single nucleotide polymorphisms. Numbering of residues is according to Abl Ia isoform. Data were collated from 27 studies published between 2001 and 2009.1-6,14,16,17,28,35,51-53,61,62,72,82-91
Methods
The panel, which comprises all the authors of this manuscript, was appointed by the European LeukemiaNet (ELN) and European Treatment and Outcome Study and included 14 among their members with a well-recognized clinical and/or research experience in CML. After critical review of relevant publications and of relevant abstracts presented at the meetings of the American Society of Hematology and of the European Hematology Association up to December 2010, the panel identified and expressed its expert opinion on key issues concerning (1) when to perform mutation analysis, (2) how to perform it, and (3) how to translate results into clinical practice.
When to look for BCR-ABL KD mutations
Imatinib first-line
Before the start of therapy: only in advanced-phase patients or in all patients?
BCR-ABL KD mutation analysis is not recommended in newly diagnosed chronic phase (CP) patients. Conversely, it can be performed in the rare cases who are in accelerated phase or blast crisis (BC) at the time of imatinib start (Table 1).
Summary of the cases in which BCR-ABL KD mutation analysis is recommended
| At diagnosis |
| Only in AP/BC patients |
| During first-line imatinib therapy |
| In case of failure |
| In case of an increase in BCR-ABL transcript levels leading to MMR loss |
| In any other case of suboptimal response |
| During second-line dasatinib or nilotinib therapy |
| In case of hematologic or cytogenetic failure |
| At diagnosis |
| Only in AP/BC patients |
| During first-line imatinib therapy |
| In case of failure |
| In case of an increase in BCR-ABL transcript levels leading to MMR loss |
| In any other case of suboptimal response |
| During second-line dasatinib or nilotinib therapy |
| In case of hematologic or cytogenetic failure |
AP indicates accelerated phase.
BCR-ABL KD mutations are not induced but simply selected by TKIs. They arise independently and may thus theoretically preexist before the start of therapy.28 In how many cases this actually happens, however, remains to be extensively addressed in large and unselected cohorts of CML patients. So far, evidence of mutations before the start of imatinib therapy has been reported only in some cases with advanced-phase disease, where the genetic instability is known to be high and the accumulation of additional genetic abnormalities is more likely. An initial report analyzed by sequencing 4 CML patients in BC who had failed to achieve any hematologic response to imatinib and found that imatinib-resistant mutations (T315I and E255K) were already detectable before imatinib start in 2 cases.6 Another study investigated the incidence of the 8 most frequent Bcr-Abl KD mutations in 66 unselected, imatinib-naive patients using a highly sensitive technique (allele-specific polymerase chain reaction [PCR]).29 No mutation was detected in the 20 patients in CP, whereas 10 of 27 accelerated phase and 5 of 19 BC patients were positive for mutations (although in several cases the mutant clone was present at very low levels).29
Should all patients on imatinib be regularly monitored for BCR-ABL KD mutations irrespective of response?
Mutation monitoring of CP patients at regular intervals during therapy is not recommended. The 6-year update of the results of the International Randomized study of Interferon and STI571 indicates that 84% of patients who achieved a complete cytogenetic response (CCyR) did not have a documented loss of CCyR.30 In line with this, very rare cases have been documented to harbor a Bcr-Abl KD mutation among patients in CCyR on imatinib. The largest study found evidence of KD mutations in 6 of 214 (< 3%) CP patients prospectively monitored by direct sequencing.31 Development of a mutation predicted for loss of CCyR; however, only 63% of the patients analyzed had received imatinib as first-line treatment, and the authors did not detail if and how many patients in this more interesting subgroup were positive for mutations and eventually relapsed. Anyway, given the low number of events occurring in CP patients who achieve a stable response on imatinib, performing mutation analysis at regular time points during therapy is worthless.
Should mutation analysis be performed only in case of failure or also in case of suboptimal response to imatinib?
Mutation analysis is recommended both in case of failure and in case of suboptimal response to imatinib (Table 1).
From a clinical standpoint, “failure” means that continuing a specific treatment is no longer appropriate because a favorable outcome is unlikely.32,33 It has been estimated that, overall, 29% of CP patients with strictly defined failure on first-line imatinib do harbor a Bcr-Abl KD mutation, although differences exist in mutation incidence across different subcategories of “failure,”34 in line with the fact that mutations are more frequently involved in acquired resistance rather than in primary resistance.35
“Suboptimal response” means that the patient may still have a substantial long-term benefit from continuing a specific treatment, but the chances of an optimal outcome are reduced.33,40 For this reason, “suboptimal responders” to imatinib may either continue on imatinib at the same dose or may become eligible for alternative approaches, including an attempt at increasing imatinib dose. We are aware that the term “suboptimal response” includes a heterogeneous group of conditions displaying heterogeneous outcome, with cytogenetic suboptimal responders showing markedly worse outcomes than molecular suboptimal responders.36-38 Overall, Bcr-Abl KD mutations have been reported in 16% of suboptimal responders, again with different subcategories exhibiting different mutation frequencies. Indeed, mutations seem to be rare in patients who do not achieve a major molecular response (MMR) by 18 months,34,39,40 although very few data are available in this regard. Nevertheless, we think that, in any scenario in which an alternative treatment approach is to be taken into consideration (as “suboptimal response” is), the knowledge of Bcr-Abl KD mutation status is an important piece of information given that a positive mutation test would support, and in some cases direct, a modification of the therapeutic strategy. Operationally, failures and suboptimal responses are identified by the absence of specific response milestones at specific time points during therapy or by the loss of a previously achieved response milestone.33 Based on the latest ELN definitions, Figure 2 may help identify these cases based on the response level achieved at each time point.

Flow chart summarizing when mutation analysis is recommended in CML patients treated with imatinib first-line. CHR indicates complete hematologic response; PCyR, partial cytogenetic response; CCA/Ph+, appearance of clonal chromosomal abnormalities in the Ph+ clone as detected by chromosome banding analysis.
Flow chart summarizing when mutation analysis is recommended in CML patients treated with imatinib first-line. CHR indicates complete hematologic response; PCyR, partial cytogenetic response; CCA/Ph+, appearance of clonal chromosomal abnormalities in the Ph+ clone as detected by chromosome banding analysis.
Should mutation analysis be performed in case of increasing BCR-ABL transcript levels? If so, which rise should trigger mutation analysis?
The panel recommends performing mutation analysis only in case the increase in BCR-ABL transcript resulted in a loss of MMR (Table 1).
In patients who are in MMR on imatinib, an increase in BCR-ABL transcript level as assessed by real-time quantitative reverse transcription (RT)–PCR might indicate a biologic change in the sensitivity of the Ph+ clone to imatinib and might herald an emerging drug resistance. As such, the ELN recommendations consider any rise in transcript level a “warning” element, requiring a more stringent and careful monitoring.32 On the other hand, fluctuations in BCR-ABL transcript level that have no clinical implication and do not anticipate a loss of response to imatinib are not infrequent; and at very low transcript levels, they can also be the result of sampling effects. Some studies have recently been published that address the issue of whether a rise in BCR-ABL transcript level may predict for loss of CCyR, thus allowing for a more timely and effective therapeutic intervention.41-44 The results of these studies were far from being concordant on the clinical significance of slight RT-PCR increases. However, 2 common points that emerged are as follows: (1) the need to have at least a confirmation of the RT-PCR results in an independent sample or, even better, a “trend” of increase resulting from 2 consecutive rises; and (2) the observation that a rise in BCR-ABL transcript level in those cases who concomitantly lose MMR is a quite reproducible predictor for loss of CCyR. Accordingly, the ELN included confirmed loss of MMR among the events defining a suboptimal response to imatinib.32,33
We can expect Bcr-Abl KD mutations to be detectable in a proportion of cases showing an increase in BCR-ABL transcripts. An initial study showed that a more than 2-fold increase in BCR-ABL transcript level was associated with the presence of a KD mutation at the time or within 3 months from the time of first rise in all the patients analyzed.45 However, this observation could not be confirmed in a subsequent independent study,46 which proposed that a rise in BCR-ABL transcript level of 2-fold or more in at least 2 consecutive evaluations, rather than a single rise, is a more reliable indicator. It is important to note that variations in the performance of local RT-PCR assays limit the general applicability of these values. For all these reasons, it is more reasonable to advise that, in patients showing an increase in BCR-ABL transcript level, only a confirmed loss of MMR be the trigger for a mutation analysis.
Dasatinib or nilotinib second-line
The second-generation TKIs dasatinib and nilotinib are approved for the treatment of imatinib-intolerant or -resistant patients. In these patients, the presence or emergence of mutations (the T315I or a few others that are known to be less sensitive to dasatinib [V299L, T315A, F317L/V/I/C]17,47-54 or nilotinib [E255K/V, Y253H, F359V/C/I]),16,51,53,54 has been reported to be a frequent cause of failure. In particular, imatinib-resistant patients who already harbor a Bcr-Abl KD mutation have been shown by several independent studies to have a higher likelihood of developing additional mutations under the selective pressure of the novel TKI, being it dasatinib or nilotinib.16,17,54
Data on the clinical value of mutation detection during the treatment with second-generation TKIs second-line are not yet sufficient to substantiate strong recommendations. However, the proposals of the ELN33 can be taken as a provisional basis to recommend that, in case of hematologic or cytogenetic failure, including no cytogenetic response at 3 months, minimal cytogenetic response (66%-95% Ph+ metaphases) at 6 months or less than partial cytogenetic response (> 35% Ph+ metaphases) at 12 months, a mutational analysis be performed. Hematologic or cytogenetic failure may be caused, or accompanied, by the appearance of a Bcr-Abl KD mutation, whose identification may be important to choose whether to switch to another TKI or proceed to allogeneic stem cell transplantation.27
Dasatinib or nilotinib first-line
The use of dasatinib or nilotinib for the first-line treatment of CML has so far been confined to the context of phase 2 and phase 3 clinical trials.55-59 In the phase 2 studies of nilotinib by the GIMEMA CML working party and by the M. D. Anderson Cancer Center, only 2 patients relapsed and progressed to BC, and they were found to harbor a T315I and an E255K mutation, respectively.55,56 The remaining published studies did not detail whether the few relapsed cases were positive for any KD mutation. Indeed, because the 2 phase 3 randomized studies57,58 have shown that, with both dasatinib and nilotinib response dynamics are more rapid, a better assessment of the value of mutation analysis in this setting requires more data and a longer follow-up.
How to look for BCR-ABL KD mutations
Direct sequencing is the method we recommend for BCR-ABL KD mutation analysis. Direct sequencing may be combined with denaturing-high performance liquid chromatography (D-HPLC) analysis, wherever this technology is available. D-HPLC is a straightforward and high-throughput tool to prescreen for sequence variations, resulting in a great reduction of the number of samples that need to be sequenced. D-HPLC and/or direct sequencing are already in use in several laboratories because they have proven a reliable method for the detection of clinically relevant Bcr-Abl KD mutations.60-65 Direct sequencing allows detection of mutations present in ≥ 20% of Ph+ cells.66 D-HPLC has a slightly higher sensitivity, but it alone does not allow characterization of the precise sequence variation underlying an abnormal elution profile. The relatively low sensitivity of these methods is not a limitation because, so far, the clinical impact of high-sensitivity BCR-ABL KD mutation detection has proven questionable. Retrospective studies in patients at diagnosis29 or in CCyR67 that tried to take advantage of highly sensitive methods, such as fluorescent allele-specific PCR, have suggested that mutations found in rare Ph+ cells are not necessarily selected; hence, their detection does not always correlate with a subsequent treatment failure because it is impossible to predict whether these cells represent a clone capable to sustain long-term hematopoiesis and effectively outcompete the unmutated one(s).68
So what… ?
Should BCR-ABL KD mutation results be used to trigger a change in therapy? If so, always or in selected cases?
When mutation analysis is performed in one of the specific cases identified in “When to look for BCR-ABL KD mutations” and with the techniques recommended in “How to look for BCR-ABL KD mutations,” a positive result represents an indication for a change in the therapeutic strategy, but the type of mutation matters. Longer and longer lists detailing the full repertoire of KD mutations have been compiled over the years based on all published studies of imatinib-resistant patients (Figure 1).18,32,66,69 For the most frequent mutations (ie, M244V, G250E, Y253F/H, E255K/V, T315I, F317L, M351T, E355G, F359V, and H396R/P), 50% inhibitory concentration (IC50) data have been published and the available clinical experience casts little doubt on a causative role in imatinib resistance. When one such mutation is detected, imatinib treatment, at least at a standard dose, is no longer advised. Many other mutations appearing in these lists are quite to very rare, no IC50 is available, and their insensitivity to imatinib, in the absence of further data, is at present rather a conjecture; we cannot rule out the possibility that they are simply “bystanders” of another resistant mechanism acting in the Ph+ clone. We must also bear in mind that constitutional single nucleotide polymorphisms have been reported in the KD (K247R; Y320C), 62,70-72 which cannot be deemed responsible for an acquired resistance. Thus, in case of a rare (or unreported) mutation with unknown IC50, a change in therapy should only be triggered by the concomitant evidence of failure or suboptimal response.
Should BCR-ABL KD mutation results be used to choose the type of second- or subsequent-line TKI? If so, always or in selected cases?
Detection of some specific mutations influences the choice of the second- or subsequent-line TKI (Table 2) because:
Summary of the most appropriate alternative therapeutic options based on the BCR-ABL KD mutation status
| T315I |
| HSCT or investigational drugs |
| V299L, T315A, and F317L/V/I/C |
| Consider nilotinib rather than dasatinib |
| Y253H, E255K/V, and F359V/C/I |
| Consider dasatinib rather than nilotinib |
| Any other mutation |
| Consider high-dose imatinib* or dasatinib or nilotinib |
| T315I |
| HSCT or investigational drugs |
| V299L, T315A, and F317L/V/I/C |
| Consider nilotinib rather than dasatinib |
| Y253H, E255K/V, and F359V/C/I |
| Consider dasatinib rather than nilotinib |
| Any other mutation |
| Consider high-dose imatinib* or dasatinib or nilotinib |
HSCT indicates hematopoietic stem cell transplantation.
No sufficient data on dose escalation available to indicate if mutations with lower IC50 values are sensitive to high-dose imatinib.
—In case of a T315I mutation, which is highly resistant to imatinib, dasatinib, and nilotinib, there are no pharmacologic opportunities other than investigational compounds in phase 1/2 clinical development.
—In case of V299L, T315A, or F317L/V/I/C mutations, nilotinib is probably more effective than dasatinib.17,47-54
—In case of Y253H, E255K/V, or F359V/C/I mutations, dasatinib is probably more effective than nilotinib.16,51,54
—In case of any other mutation, dasatinib and nilotinib are likely to be similarly effective.
Obviously, dasatinib and nilotinib exhibit different IC50 values (and different fold changes in IC50 with respect to unmutated Bcr-Abl) for each individual Bcr-Abl mutant form as a consequence of their different chemical structures and binding modes. When a given mutation is detected, it is thus tempting to let the TKI choice result from the comparison of the IC50 values for that mutant, if available, selecting the inhibitor with the lower IC50. However, we recommend clinicians to bear in mind that IC50 tables are an imperfect tool to guide optimal selection of the second- or subsequent-line TKI. On one hand, a certain degree of correlation between dasatinib and nilotinib IC50 for a specific mutant in vitro and clinical responses in patients harboring the same mutant in vivo exists, in that those harboring mutations with higher IC50 values had lower hematologic and cytogenetic response rates than those harboring mutations with lower IC50 values.16,17,53 On the other hand, for some mutations, there is little concordance between the IC50 values reported by different studies. Tables 3 and 4 report all the cellular IC50 (and the fold changes in IC50 with respect to unmutated Bcr-Abl) obtained in BaF3 cellular systems in 10 published studies.6,11,24,26,47,73-77 When comparing the values for the same mutant and the same inhibitor across different reports, up to 10-fold differences can be noticed; this is most probably a reflection of the different experimental methods and conditions used to derive the IC50 but makes it difficult, for several mutations, to draw firm predictions on their actual degree of sensitivity to a TKI. In addition, IC50 values are derived using a cell line as a model, and this does not allow accounting for a spectrum of factors determining the effective intracellular drug concentrations achievable in humans, such as absorption, metabolism, distribution to target compartments, transport, and excretion. These factors are also subject to a certain degree of interindividual variability. Thus, physicians should be aware that an IC50-based prediction of which one, between dasatinib and nilotinib, will be more effective could not always lead to the expected clinical responses.
Cellular IC50 (nM) of imatinib (IM), nilotinib (NI), and dasatinib (DA) and fold increase with respect to the IC50 for wild-type (WT) Bcr-Abl of the M244V, L248V, G250E, Q252H, Y253H, Y253F, E255K, E255V, E279K, and V299L mutant forms
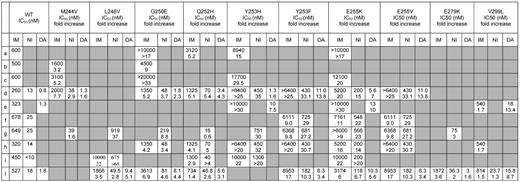
Data were collated from 10 published studies. a, Shah et al6 ; b, Corbin et al11 ; c, Azam et al73 ; d, O'Hare et al24 ; e, Burgess et al47 ; f, Manley et al74 ; g, Weisberg et al26 ; h, Bradeen et al75 ; i, Ray et al76 ; and l, Redaelli et al.77 Mouse lymphoblastoid cells (BaF3) transfected with p210BCR-ABL were used in all the studies. The IC50 was calculated with different methods, namely, trypan blue exclusion,6,26,47,76 MTT,11 WST-1,73 or MTS24,76 colorimetric assays, ATP-lite fluorescent assay,26,74 and tritiated thymidine incorporation.77 In one study75 the method was not detailed. IC50 was assessed after 48,6,11 60,73 70,26,74 or 7224,47,76 hours of incubation with the inhibitors. In the other studies,75,77 this information was not provided.
Cellular IC50 (nM) of imatinib (IM), nilotinib (NI), and dasatinib (DA) and fold increase with respect to the IC50 for wild-type (WT) Bcr-Abl of the F311L, T315I, F317L, M351T, F359V, V379I, L384M, L387M, H396R, H396P, and F486S mutant forms
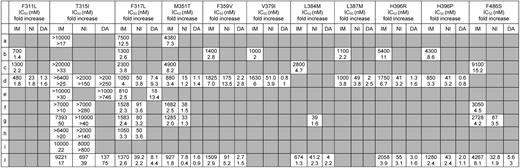
Data were collated from 10 published studies. As far as the remaining mutations are concerned, IC50 values (and fold changes in IC50), when available, are not included in these tables because they have been assessed in single studies: D276G, 1147nM (2.2), 35.3nM (2), and 2.6nM (1.4) for IM, NI, and DA, respectively;77 T315A, 760nM (2.4) and 125nM (93) for IM and DA, respectively;47 F317V, 500nM (1.6) and 350nM (25) for IM and NI, respectively;75 F317C, 1200nM (3.8) for IM;75 and E355G, 2380nM (4) for IM.6
Conclusions
Here we aimed to provide recommendations to clinicians on how to best integrate BCR-ABL KD mutation analysis in the routine management of CML patients. We acknowledge that the literature is still not comprehensive enough to base all our recommendations on sound evidence. The reader should therefore be warned that these are mostly based on the expert opinion of the members of this panel.
The cases in which a BCR-ABL KD mutation analysis is recommended are summarized in Table 1. This is mainly intended for newly diagnosed patients receiving imatinib first-line. However, results of imatinib second-line in CP patients78-80 do not differ so significantly from those of imatinib first-line to require specific recommendations, except for a minority of patients with longer disease history who may require a more thorough, case-by-case evaluation. The recommended method for BCR-ABL KD mutation analysis is direct sequencing (which may be preceded by D-HPLC screening). More sensitive strategies should remain confined to a research area because they have so far failed to soundly demonstrate that “earlier is better,” and their results cannot represent a reliable trigger for therapeutic intervention. Although the choice of the second- or subsequent-line strategy must result from a decision algorithm, including several factors, such as patient history, risk factors, and comorbidities, detection of some specific mutations (the T315I above all) should be part of this algorithm, as summarized in Table 2.
Bcr-Abl KD mutations are the most extensively investigated mechanism of resistance to imatinib, but they are not the only one. Actually, the frequency by which mutations have been implicated in imatinib resistance is different in the different phases of CML,35,81 ranging from 25% to 30% of early CP patients on first-line imatinib to approximately 70% to 80% of BC patients. In addition, Bcr-Abl KD mutations can more commonly be detected in cases showing acquired resistance than in cases with primary resistance.35 In mutation-negative patients, other resistance mechanisms have been shown, or hypothesized, to intervene.18 It is also conceivable that, in a proportion of cases, more than one factor may cooperate to determine the resistance phenotype. We cannot even exclude that, in some patients, mutations may be simple “bystanders.” This is unlikely in the presence of mutations highly insensitive to imatinib and for which a molecular mechanism of resistance has been posited (eg, T315I, P-loop mutations, F359V) but might be the case for those less frequent mutations for which we have more limited knowledge. Therefore, we recommend a change in therapy always to be triggered by concomitant evidence of failure or suboptimal response to imatinib. It is also important to bear in mind that, whatever the actual contribution of a mutation to the resistant phenotype is, its presence by itself should not be overlooked: mutations may be a sign of genetic instability, and genetic instability is the engine of disease evolution toward a more aggressive phenotype.
Optimization of CML treatment is a continuous process. Knowledge is still accumulating, and the therapeutic scenario is still evolving. Nilotinib and dasatinib have just become available for newly diagnosed CML. In case of wider use of these TKIs in the first-line setting, the clinical impact of Bcr-Abl KD mutations would probably change profoundly. These recommendations might be perceived as provisional under some aspects, but we think they can provide a valuable aid to clinicians in advising patients and managing current CML treatment.
Acknowledgments
European LeukemiaNet is supported by the European Union, Sixth Framework Programme (contract no. LSHC-CT-2004-503216), and Novartis Oncology Europe through the European Treatment and Outcome Study for CML. ELN provided the scientific platform for this study but no direct financial support.
Authorship
Contribution: S.S. drafted the manuscript, tables, and figures; and all authors conceived and designed the study, collected and assembled data, analyzed and interpreted data, critically revised the manuscript for important intellectual content, and gave final manuscript approval.
Conflict-of-interest disclosure: S.S. received speaker fees and travel support from Novartis and Bristol-Myers Squibb. A.H. received research funding and honoraria from Novartis and Bristol-Myers Squibb. F.E.N. received consultancy and speaker fees from Novartis and Bristol-Myers Squibb. T.L. received honoraria from Novartis and Bristol-Myers Squibb and research funding from Novartis. G.S. received consultancy and speaker fees from Novartis and Bristol-Myers Squibb. F.P. received consultancy and speaker fees from Novartis and Bristol-Myers Squibb and research funding from Novartis. M.C.M. received research funding from Novartis, honoraria from Novartis and Bristol-Myers Squibb, and travel support from Novartis and Bristol-Myers Squibb. G.R. received consultancy and speaker fees from Novartis and Bristol-Myers Squibb. K.P. received honoraria and research funding from Bristol-Myers Squibb and Novartis. M.B. received consultancy and speaker fees from Novartis and Bristol-Myers Squibb. N.C.P.C. received honoraria and speaker fees from Novartis and Bristol-Myers Squibb. G.M. received consultancy and speaker fees from Novartis and Bristol-Myers Squibb and consultancy for Pfizer. The remaining authors declare no competing financial interests.
Correspondence: Simona Soverini, S. Orsola-Malpighi Hospital, Via Massarenti 9, 40138 Bologna, Italy; e-mail: simona.soverini@tin.it.
