

Abstract
Abstract 4855
Sickle cell disease (SCD) results from a point mutation in the beta globin gene that leads to the replacement of glutamic acid with a valine at the sixth position of the beta globin chain of hemoglobin. Children with SCD experience delayed skeletal maturation and more than 70% of adult SCD patients' exhibit osteopenia or osteoporosis. The underlying mechanisms responsible are unclear; however in a number of disorders inflammation is established to drive bone loss. Importantly, persistent immune-activation in SCD leads to a chronic inflammatory state that may be pertinent. In this study we characterized the skeletons of four month old Berkeley SCD mice, an animal model that recapitulates key features of SCD including inflammation. We show that as in children with SCD, the skeletons of SCD mice have significantly reduced bone mineral density (BMD). Micro-computed tomography shows that homozygous sickle cell mice were also significantly denuded in cancellous and cortical bone volumes, a consequence of an enhanced pro-osteoclastogenic bone microenvironment. Similarly to animal models of postmenopausal osteoporosis we found a significant increase in T cell activation state and TNFa production in SCD mice, and an elevated expression of receptor activator of NF-kB ligand (RANKL), the key osteoclastogenic cytokine in the bone marrow. Finally, we show a significantly elevated number of monocytes (early osteoclast precursors) that likely further amplifies osteoclastogenesis in the context of elevated RANKL in SCD. Taken together our data validate the Berkeley sickle mouse as a suitable model to explore the mechanisms of bone loss in SCD, and suggest a disruption in the immuno-skeletal interface resulting from chronic inflammation as key to bone loss in SCD.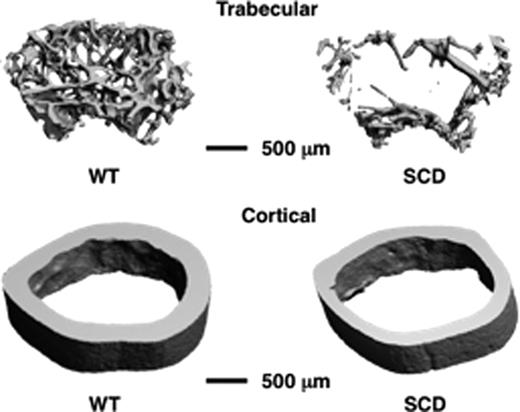

Disclosures:
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal