

Protein disulfide isomerase (PDI) catalyzes the oxidation reduction and isomerization of disulfide bonds. We have previously identified an important role for extracellular PDI during thrombus formation in vivo. Here, we show that endothelial cells are a critical cellular source of secreted PDI, important for fibrin generation and platelet accumulation in vivo. Functional PDI is rapidly secreted from human umbilical vein endothelial cells in culture upon activation with thrombin or after laser-induced stimulation. PDI is localized in different cellular compartments in activated and quiescent endothelial cells, and is redistributed to the plasma membrane after cell activation. In vivo studies using intravital microscopy show that PDI appears rapidly after laser-induced vessel wall injury, before the appearance of the platelet thrombus. If platelet thrombus formation is inhibited by the infusion of eptifibatide into the circulation, PDI is detected after vessel wall injury, and fibrin deposition is normal. Treatment of mice with a function blocking anti-PDI antibody completely inhibits fibrin generation in eptifibatide-treated mice. These results indicate that, although both platelets and endothelial cells secrete PDI after laser-induced injury, PDI from endothelial cells is required for fibrin generation in vivo.
Introduction
A considerable body of evidence implicates the oxidation state of labile disulfide bonds in critical hemostatic proteins in regulating the process of thrombus formation.1 The oxidation state of these bonds is regulated by an enzyme(s) of the thiol isomerase family. Thiol isomerases, including protein disulfide isomerase (PDI), while containing endoplasmic reticulum retention signals, are found extracellularly. Among the cells that secrete PDI and display the enzyme on their surface are platelets and endothelial cells.2,,,,–7 The importance of thiol-disulfide balance for platelet function has long been recognized. For example, reduced glutathione and cysteine inhibit platelet aggregation induced by several agonists, while dithiothreitol and β-mercaptoethanol promote aggregation.4 PDI likely plays an important role in maintaining this balance. The levels of both PDI and ERp5, another member of the PDI family, on the platelet surface increase significantly upon agonist stimulation.4,6 PDI has been implicated in αIIbβ3 and α2β1 activity,8,9 and glycoprotein Ib α expresses one or more free thiols on the activated platelet surface, but not on resting platelets.4 Inhibitory anti-PDI antibodies or bacitracin, a nonspecific inhibitor of thiol isomerases, inhibit platelet activation in vitro, suggesting that αIIbβ3-dependent platelet aggregation and secretion require thiol isomerases.10 PDI may play a role in the de-encryption of tissue factor.11,–13
In contrast, there is less information to support potential roles of extracellular thiol isomerases in the function of endothelial cells. Endothelial cells in culture secrete PDI, which then is bound to the cell surface.5 A novel thiol isomerase that appears to be endothelial cell specific, EndoPDI or ERp46, has been reported.14 Recent evidence indicates that the protein disulfide isomerases, ERp46 and ERp57, are present in endothelial cell plasma membrane preparations.15 Endothelial cells in culture secrete an activity that reduces the size of very large multimers of von Willebrand factor (VWF).16 This activity appears to be independent of the proteolysis of VWF by ADAMTS13 and is inhibited by thiol blocking reagents. The VWF reductant secreted from endothelial cells has been identified as thrombospondin-1.17 A functional role for extracellular thiol isomerases on endothelial cell activation has not been explored.
We and others have recently determined that PDI plays a significant role in thrombus formation in vivo.18,19 Using intravital fluorescence microscopy after laser-induced vessel wall injury in mouse cremaster muscle arterioles, we determined that there is a time-dependent increase in PDI at the site of thrombus formation after injury. Infusion of bacitracin or a blocking monoclonal antibody to PDI into the circulation inhibited both platelet thrombus formation and fibrin generation.18 Although the presence of PDI in plasma has been controversial20,21 we did not detect significant amounts of PDI in human or mouse plasma (vide infra). Hence, the PDI that plays a role in thrombus formation is likely contributed by cells activated at the site of thrombus formation.
Fibrin deposition is normal in our laser thrombosis model in mice lacking the thrombin receptor PAR4.22 Although there is initial platelet accumulation after laser-induced arteriolar injury in these mice, platelet accumulation is minimal, and the platelets in the juxtamural thrombus that forms in Par4−/− mice are activated only after a long delay. These results suggest that platelets within the juxtamural thrombi cannot support fibrin generation.22 Inhibition of PDI eliminated fibrin accumulation in Par4−/− mice, indicating that the enzyme plays a necessary role in thrombin generation in this model. Inhibition of PDI also eliminated the small juxtamural thrombus usually formed in Par4−/− mice. Analysis of the kinetics of PDI expression after laser-induced vessel injury shows the appearance of PDI before platelet accumulation.18 Thus, there must be an alternative to the activated platelet as a source of PDI at the site of laser-induced vessel wall injury. Because endothelial cells lining the arteriolar wall are the direct target of the laser injury in this thrombosis model, we have explored the role of endothelial PDI in thrombus formation. Here, we show that endothelial PDI is required for fibrin generation, whereas platelet PDI contributes to the total amount of thrombus-associated PDI, but is not required.
Methods
Animals
C57BL/6J WT mice were obtained from Jackson Laboratory. The Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee approved all animal care and experimental procedures.
Antibodies and reagents
Rat anti–mouse CD41 antibody (clone MWReg30) and R300 platelet-depleting antibodies against mouse GP1bα were purchased from Emfret. Fab fragments from the CD41 antibody were generated using the ImmunoPure Fab Preparation Kit (Pierce Biotechnology). Anti-fibrin antibody (clone NYBT2G1) and Nycodenz density-gradient medium were purchased from Accurate Chemicals. Anti-PDI antibody RL90 was from Abcam. Polyclonal anti-PDI antibody was from Sigma-Aldrich. Anti-VWF (Chemicon), antialkaline phosphatase (clone 4H1; Abcam), and anti-sarcoplasmic and endoplasmic reticulum calcium ATPase 2b (SERCA 2b; clone 2A7-A1; ABR Affinity Bioreagents) antibodies were used as markers for cell localization studies. Growth-related oncogene-α (Gro-α)-polyclonal antibody was purchased from Peprotech. Antibodies and Fab fragments were labeled with Alexa 488 or Alexa 647 (Invitrogen). Alexa 488– or Alexa 647–labeled secondary antibodies, Fluo-4 and Rhod-2, were purchased from Invitrogen. Human α-thrombin, calcium ionophore A23187, histamine, bovine insulin, and phorbol myristate acetate (PMA) were purchased from Sigma-Aldrich. Human umbilical vein endothelial cells (HUVECs), Medium 200, and low-serum growth supplement were obtained from Cascade Biologics. Eptifibatide was purchased from Schering-Plough.
Cell culture and immunoblotting
HUVECs were cultured in Medium 200 supplemented with low-serum growth supplement. Confluent cells from passages 2-5 were used. Cells were washed with phosphate-buffered saline (PBS), rinsed with serum-free medium for 10 minutes, and incubated overnight in fresh serum-free medium. To induce regulated secretion of PDI (ie, acute release), these cells were incubated for the indicated times with the following: 1 NIH U/mL of human α-thrombin, 10μM calcium ionophore A23187, 0.1mM histamine, or 100 ng/mL PMA. The medium was collected at specific time points. PDI in serum-free media from resting and activated HUVECs was detected by immunoblotting. The amount of PDI secreted from resting and activated cells was quantitated by comparison to the density units of known amounts of purified recombinant human PDI.
PDI activity
PDI activity was determined in the medium of thrombin-activated endothelial cells using the insulin transhydrogenase assay.23 Bovine insulin (1 mg/mL) in 250 μL of 50mM Tris-HCl buffer (pH 7.5) was incubated with 50 μL of HUVEC medium in the presence or absence of the monoclonal RL90 antibody or an isotype-matched IgG2a control. The turbidometric assay of insulin disulfide reduction by PDI was initiated with 3 mL of 100mM dithiothreitol, and the reaction was monitored at 650 nm over 60 minutes.
Intravital microscopy
Intravital video microscopy of the cremaster muscle microcirculation was performed as previously described.24,25 The intravital fluorescence microscopy system has previously been described in detail.26 Confocal intravital images were obtained using a Yokagawa CSU-X1 confocal head with 1-μm z-stacks controlled by a piezoelectric driver from Physik Instrumente. Digital images were captured with a Cooke Sensicam charge-coupled device camera (The Cooke Corporation) connected to a VS4-1845 Image Intensifier GEN III (Video Scope International).
Laser-induced injury
Injury to an arteriolar (30-50-μm diameter) vessel wall was induced with a Micropoint Laser System (Photonics Instruments) focused through the microscope objective, parfocal with the focal plane, and tuned to 440 nm and the dye cell containing 5mM coumarin in methanol.27 Data were captured digitally from 2 fluorescence channels: 488/520 nm and 647/670 nm. Data acquisition was initiated both before and after the pulsing of the laser for each injury. The microscope system was controlled and images were analyzed using the software package Slidebook Version 4.2 or higher (Intelligent Imaging Innovations).
Image analysis
For each thrombus generated, a rectangular mask was defined that included a portion of the vessel upstream of the site of injury. The maximum fluorescence intensity of the pixels contained in this mask was extracted for all frames (pre- and postinjury) for each thrombus. The mean value calculated from the maximal intensity values in the mask for each frame was determined and used as the background value. Finally, for each frame, the integrated fluorescence intensity was calculated as per the following equation:
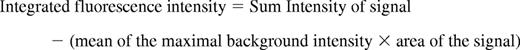
This calculation was performed for all frames in each thrombus and plotted vs. time to provide the kinetics of thrombus formation. For multiple fluorescence channels, calculations of background were made independently for each channel. The data from 25-30 thrombi were used to determine the median value of integrated fluorescence intensity to account for the variability of thrombus formation at any given set of experimental conditions.
Density gradient centrifugation
Confluent HUVECs were washed in PBS and scraped into 2 mL of homogenization buffer (5mM Tris-HCl, 220mM sucrose, 0.001% Tween 80, pH 7.4) and protease inhibitor cocktail. All steps were performed on ice unless otherwise indicated. Cells (2 × 107) were lysed by 3 freeze-thaw cycles at −140°C and 37°C. The cell lysate was centrifuged at 800g for 5 minutes at 4°C to remove cell debris, and the supernatant was retained for fractionation. Next, 2 mL of cell lysate was layered on top of a Nycodenz density gradient that had been equilibrated overnight at 4°C. The 11 step gradient, prepared in a 14 mL ultracentrifuge tube, contained 1 mL each of Nycodenz from 30% to 5% (wt/vol in homogenization buffer) in 2.5% increments layered over a cushion of 1 mL of 35% Nycodenz (wt/vol in 5mM Tris-HCl, pH 7.4). The gradient was centrifuged for 150 minutes at 40 000 rpm (202 000gav) in a Beckman LE-80K ultracentrifuge equipped with a SW-40 rotor, at 4°C. Subsequently, 1 mL fractions of the gradient were collected from the bottom of the tube and stored at −20°C. The density of each fraction was determined and ranged from 1.02 to 1.19 g/mL.28
Immunonanogold electron microscopy
HUVECs were resuspended in 5mM EDTAPBS. The cell suspension was added to a microfuge tube containing an equal volume of 2× fixative (4% paraformaldehyde in 0.1M sodium phosphate buffer; pH 7.4). After fixing for 2 hours, the cells were pelleted by centrifugation. The cell pellet was infiltrated with 2.3M sucrose for 15-30 minutes at room temperature and cut into small blocks, which were mounted on pins and frozen in liquid nitrogen. Ultrathin cryosections of 60-70 nm were obtained by cutting the blocks at −120°C with a Reichert Ultracut S ultracryomicrotome (Leica) and were picked up in a 1:1 mixture of 2% methylcellulose and 2.3M sucrose. The cryosections were transferred to formvar-carbon–coated copper grids and floated on PBS. Grids were floated on drops of 1% bovine serum albumin (BSA) for 10 minutes to block nonspecific labeling. For the single labeling, cryosections were sequentially incubated for 30 minutes at room temperature with the RL90 mouse anti-PDI antibody RL90, then protein A–coated colloidal gold particles (10 nm) in 1× PBS with 1% BSA. For double labeling, cryosections were first labeled with RL90 and protein A–coated colloidal gold particles (5 nm) and postfixing in 1% glutaraldehyde. The sections were then blocked with 0.2M glycine to neutralize free aldehyde groups for 15 minutes and incubated with the anti-SERCA2b antibody for 30 minutes. Protein A–coated colloidal gold particles (15 nm) were used to detect SERCA2b in endoplasmic reticulum. The cells were finally postfixed in 1× PBS containing 1% glutaraldehyde and contrasted with a 1:9 mixture of 3% uranyl-acetate and 2% methylcellulose. The grids were examined in a Tecnai G2 Spirit BioTWIN transmission electron microscope, and images were recorded with an AMT 2k charge-coupled device camera.
Endothelial cell stimulation and immunostaining
Monolayers of HUVECs were cultured on 0.1% gelatin-coated coverslips until they reached 80% confluence. For laser activation, cells were grown on photoetched coverslips (Bellco). Before laser stimulation, endothelial cells were loaded with Fluo-4, as per the manufacturer's instruction. The cells were then immersed in recalcified plasma supplemented with corn-trypsin inhibitor (100 μg/mL; Hematologic Technologies) and a specific PDI inhibitory antibody RL90 or an isotype control antibody. Designated areas on the etched coverslip were then activated with the Micropoint Laser System, and calcium mobilization was recorded. The reaction with plasma was stopped after 15 minutes with 20mM ethylenediaminetetraacetic acid, and the cells were immediately fixed in 3% paraformaldehyde for 5 minutes. Postfixation, the cells were washed with 1× PBS and stained with anti-fibrin antibody (clone 59D8) or an isotype control antibody. For studies involving cellular localization of PDI, fixed cells were permeabilized with 1% Triton X-100 for 5 minutes and blocked with 5% normal goat serum. Specific antibodies to PDI (monoclonal RL90), VWF (polyclonal anti-VWF), or SERCA 2b (clone 2A7-A1) were used. An idiotype-matched antibody was used as a control, followed by appropriate Alexa 488– or Alexa 647–labeled secondary antibodies. For double labeling, the primary and secondary antibody cycle was repeated with a second set of antibodies. Images were captured using a Roper Coolsnap HQ camera.
Results
Regulated secretion of PDI from endothelial cells
PDI appears at sites of thrombus formation in an in vivo mouse laser-induced injury model of thrombosis, and we have established that platelets are one source of PDI at the injury site.18 To determine whether endothelial cells are a potential source of PDI during thrombus formation, we examined the secretion of PDI from HUVECs in culture. PDI levels were very low, as measured by Western blotting of serum-free medium collected after overnight culture of HUVECs (Figure 1A lane 1). Activation of HUVECs by thrombin (Figure 1A lane 2) or the calcium ionophore, A23187 (Figure 1A lane 3), resulted in PDI secretion from HUVECs. To demonstrate that the PDI in the medium from activated HUVECs is due to the presence of soluble PDI and not PDI-bearing microparticles shed from HUVECs, particulates in the conditioned medium were sedimented by centrifugation for 2 hours at 100 000g and proteins in the supernatant were detected by Western blotting with anti-PDI antibody. The postcentrifugation supernatant contained essentially all of the PDI observed in the medium of endothelial cells activated by thrombin or calcium ionophore (data not shown). The release of PDI as a function of time from HUVECs activated with 1 U of thrombin is shown in Figure 1B. PDI was released and detectable in the culture medium from activated HUVECs within 5 minutes after stimulation with thrombin. The amount of accumulated PDI detected in the culture medium increased over a period of 30 minutes (Figure 1B-C), with a corresponding reduction in the PDI detected in cell lysates from the thrombin-activated HUVECs (Figure 1B). Approximately 6 ng of PDI was detected in the media from 5 × 106 cells after 30 minutes of stimulation with thrombin. This corresponds to approximately 1.2 fg of PDI secreted per HUVEC. Approximately half of the secreted PDI was detected in the media within the first 5 minutes after thrombin stimulation, suggesting a rapid secretory response of HUVECs to agonist-induced activation. Similar kinetics of PDI secretion from HUVECs were observed after stimulation with PMA (100 ng/mL). Histamine (0.1mM) stimulation led to lower levels of PDI secretion with a slower time course. The secreted PDI was functionally active, as determined by the insulin transhydrogenase assay. The activity was inhibited with the function blocking anti-PDI antibody RL90 (Figure 1D).

Agonist-induced PDI secretion from HUVECs. PDI secretion from HUVECs into serum-free cell culture medium after overnight culture of resting or activated cells was detected by sodium dodecyl sulfate–electrophoresis of the medium followed by immunoblotting with anti-PDI antibodies. (A) PDI in culture medium (50 μL from confluent monolayer ∼ 1 × 106 cells) detected with the monoclonal anti-PDI antibody, RL90, at 1 μg/mL. Lane 1, conditioned media from resting HUVECs; lane 2, conditioned medium from HUVECs activated with 1 U/mL thrombin; lane 3, conditioned medium from HUVECs activated with 10μM A23187 calcium ionophore. (B) Immunoblot showing PDI secretion after thrombin stimulation from 5 × 106 HUVECs over a period of 30 minutes. Top panel, medium from thrombin-activated HUVECs. Bottom panel, cell lysates from the corresponding activated cells. (C) Time course study of PDI release from 5 × 106 HUVECs after stimulation with (■) 100 ng/mL PMA, (♦) 1 U/mL thrombin, or (▴) 0.1mM histamine. (D) PDI activity was measured by the insulin transhydrogenase assay in the conditioned media from 5 × 106 thrombin-activated HUVECs (mean of 3 experiments) in the presence (unfilled) or absence (filled) of 5 μg/mL monoclonal inhibitory antibody RL90. Circles, PDI activity secreted from cultured HUVECs; triangles, activity of recombinant human PDI (1 μg/mL).
Agonist-induced PDI secretion from HUVECs. PDI secretion from HUVECs into serum-free cell culture medium after overnight culture of resting or activated cells was detected by sodium dodecyl sulfate–electrophoresis of the medium followed by immunoblotting with anti-PDI antibodies. (A) PDI in culture medium (50 μL from confluent monolayer ∼ 1 × 106 cells) detected with the monoclonal anti-PDI antibody, RL90, at 1 μg/mL. Lane 1, conditioned media from resting HUVECs; lane 2, conditioned medium from HUVECs activated with 1 U/mL thrombin; lane 3, conditioned medium from HUVECs activated with 10μM A23187 calcium ionophore. (B) Immunoblot showing PDI secretion after thrombin stimulation from 5 × 106 HUVECs over a period of 30 minutes. Top panel, medium from thrombin-activated HUVECs. Bottom panel, cell lysates from the corresponding activated cells. (C) Time course study of PDI release from 5 × 106 HUVECs after stimulation with (■) 100 ng/mL PMA, (♦) 1 U/mL thrombin, or (▴) 0.1mM histamine. (D) PDI activity was measured by the insulin transhydrogenase assay in the conditioned media from 5 × 106 thrombin-activated HUVECs (mean of 3 experiments) in the presence (unfilled) or absence (filled) of 5 μg/mL monoclonal inhibitory antibody RL90. Circles, PDI activity secreted from cultured HUVECs; triangles, activity of recombinant human PDI (1 μg/mL).
PDI is distributed in different cytosolic compartments in resting and active endothelial cells
Because endothelial cells secrete PDI upon agonist stimulation, we performed density-gradient centrifugation to determine the distribution of PDI in cellular fractions in resting and activated endothelial cells. Sodium dodecyl sulfate gel electrophoresis and Western blot analysis of density gradient fractions from lysates of resting endothelial cells showed that PDI was detected as a single, almost symmetrical peak, with a maximum at density 1.087 g/mL and a range of 1.06-1.12 g/mL (Figure 2A-B). Both the peak of PDI activity, as measured by the insulin transhydrogenase assay, and the peak of PDI antigen were found at the same position in the gradient (Figure 2B fraction 9). The correlation between PDI activity and antigen in 14 fractions in 2 repeat experiments was > 0.97. The peak density for the endoplasmic reticulum marker SERCA2b29 was found at density 1.099 g/mL, and SERCA2b was distributed among fractions of density 1.059-1.121 g/mL. This distribution was very similar to that of PDI in the resting cells. Analysis of lysates from activated endothelial cells showed that PDI antigen and activity were present in fractions of a broader density range from 1.045-1.12 g/mL. Alkaline phosphatase, a marker for the plasma membrane, was detected around density 1.03-1.08 g/mL (Figure 2C), suggesting that PDI may be located on the plasma membrane of activated endothelial cells. PDI was not found in fractions containing the Weibel-Palade body marker, VWF, which was detected at densities between 1.11 and 1.17 g/mL.

PDI distribution in resting and activated endothelial cells by gradient centrifugation. Organelles in a HUVEC lysate were separated by centrifugation on a Nycodenz density gradient. Percentages refer to percentage per fraction of total activity or antigen recovered in the 14 fractions of the gradient. The numbers 1-14 of the fractions are displayed on the x-axis. (A) Immunoblots of fractions 1-14 (higher to lower density, right to left) using the monoclonal PDI antibody, RL90, in fractions from unactivated (top panel, PDI) or thrombin-activated (second panel, PDI+) HUVECs. Middle and bottom panels show immunoblots for distribution of markers for endoplasmic reticulum, SERCA 2b; Weibel-Palade bodies, VWF; or plasma membrane, alkaline phosphatase, with (+) or without thrombin activation of cells. The numbers on the immunoblots refer to the density-gradient fractions. (B) The percentage per fraction of PDI antigen (—) or PDI enzymatic activity (- - -) are shown as the average of 2 experiments. The open circles (○) indicate fractions from unactivated HUVECs, while closed circles (•) show fractions from thrombin-activated HUVECs. The mean density of the fractions is indicated by the gray dashed line. (C). The average percentage per fraction from 2 experiments for markers, alkaline phosphatase (▴), SERCA2b (•), or VWF (■).
PDI distribution in resting and activated endothelial cells by gradient centrifugation. Organelles in a HUVEC lysate were separated by centrifugation on a Nycodenz density gradient. Percentages refer to percentage per fraction of total activity or antigen recovered in the 14 fractions of the gradient. The numbers 1-14 of the fractions are displayed on the x-axis. (A) Immunoblots of fractions 1-14 (higher to lower density, right to left) using the monoclonal PDI antibody, RL90, in fractions from unactivated (top panel, PDI) or thrombin-activated (second panel, PDI+) HUVECs. Middle and bottom panels show immunoblots for distribution of markers for endoplasmic reticulum, SERCA 2b; Weibel-Palade bodies, VWF; or plasma membrane, alkaline phosphatase, with (+) or without thrombin activation of cells. The numbers on the immunoblots refer to the density-gradient fractions. (B) The percentage per fraction of PDI antigen (—) or PDI enzymatic activity (- - -) are shown as the average of 2 experiments. The open circles (○) indicate fractions from unactivated HUVECs, while closed circles (•) show fractions from thrombin-activated HUVECs. The mean density of the fractions is indicated by the gray dashed line. (C). The average percentage per fraction from 2 experiments for markers, alkaline phosphatase (▴), SERCA2b (•), or VWF (■).
PDI is localized to a distinct cytosolic compartment in HUVECs
The rapid release of PDI from HUVECs in response to thrombin, PMA, and histamine suggests that PDI is stored in a distinct compartment for regulated secretion. However, the results of cell fractionation experiments indicated that PDI is not stored in Weibel-Palade bodies, the most prominent storage granules of endothelial cells. We used confocal and electron microscopy to further establish the cellular location of PDI. As anticipated, PDI was partially colocalized with the endoplasmic reticulum resident protein SERCA2b in paraformaldehyde-fixed HUVECs (Figure 3A). Punctate PDI staining was also observed in the cytosol of HUVECs, but, as in the cell fractionation studies, the PDI-containing storage granules did not colocalize with VWF in Weibel-Palade bodies (Figure 3B). Distinct chemokine-containing granules have been described in endothelial cells.30,31 Coimmunostaining for PDI and GRO-α, a chemokine secreted from these small granules, showed partial colocalization of these proteins (Figure 3C). Similar partial colocalization of PDI with monocyte chemo-tactic protein-1 was observed in HUVECs (data not shown). Monocyte chemotactic protein-1 has previously been shown to colocalize with GRO-α in small secretory granules.30,31

Cellular distribution of PDI in HUVECs. Intracellular localization of PDI was detected by immunostaining of fixed cultured HUVECs with monoclonal antibody RL90. (A) Simultaneous immunostaining of cells for PDI and SERCA2b in HUVECs indicates that these 2 proteins are colocalized in the endoplasmic reticulum. In addition, PDI is observed in granules distinct from the endoplasmic reticulum. Alexa 647–labeled anti-SERCA2b, red; Alexa 488–labeled RL90, green; colocalization, yellow. (B) Simultaneous immunostaining of cells for PDI and VWF indicates that PDI is not stored in Weibel Palade bodies. Alexa 647–labeled goat anti–rabbit IgG was used as a secondary antibody to detect VWF, red; Alexa 488–labeled RL90, green; colocalization, yellow. (C) Simultaneous immunostaining of chemokine Gro-α and PDI shows partial colocalization in small cytoplasmic granules. Original magnification in all panels ×60. Insets show high magnification (×100) of framed areas. (D) Immunogold labeling for PDI (10-nm gold particles) in HUVECs showed PDI in endoplasmic reticulum–related tubulovesicular structures and in small moderately electron-dense granules of approximately 100-150-nm diameter. WPB, Weibel-Palade bodies; ER, endoplasmic reticulum, SG, moderately electron-dense secretory granules (×99 000). (E) PDI and SERCA2b are colocalized in the endoplasmic reticulum, as indicated by immunogold staining. Only PDI is detected in secretory granules (×99 000). F) No plasma membrane–associated signal is detected in resting HUVECs (×119 000). (G) PDI, but not SERCA2b, is bound to the plasma membrane in activated HUVECs (×119 000). PM, plasma membrane. Panels E-G: PDI, 5-nm gold particles; SERCA2b, 15-nm gold particles.
Cellular distribution of PDI in HUVECs. Intracellular localization of PDI was detected by immunostaining of fixed cultured HUVECs with monoclonal antibody RL90. (A) Simultaneous immunostaining of cells for PDI and SERCA2b in HUVECs indicates that these 2 proteins are colocalized in the endoplasmic reticulum. In addition, PDI is observed in granules distinct from the endoplasmic reticulum. Alexa 647–labeled anti-SERCA2b, red; Alexa 488–labeled RL90, green; colocalization, yellow. (B) Simultaneous immunostaining of cells for PDI and VWF indicates that PDI is not stored in Weibel Palade bodies. Alexa 647–labeled goat anti–rabbit IgG was used as a secondary antibody to detect VWF, red; Alexa 488–labeled RL90, green; colocalization, yellow. (C) Simultaneous immunostaining of chemokine Gro-α and PDI shows partial colocalization in small cytoplasmic granules. Original magnification in all panels ×60. Insets show high magnification (×100) of framed areas. (D) Immunogold labeling for PDI (10-nm gold particles) in HUVECs showed PDI in endoplasmic reticulum–related tubulovesicular structures and in small moderately electron-dense granules of approximately 100-150-nm diameter. WPB, Weibel-Palade bodies; ER, endoplasmic reticulum, SG, moderately electron-dense secretory granules (×99 000). (E) PDI and SERCA2b are colocalized in the endoplasmic reticulum, as indicated by immunogold staining. Only PDI is detected in secretory granules (×99 000). F) No plasma membrane–associated signal is detected in resting HUVECs (×119 000). (G) PDI, but not SERCA2b, is bound to the plasma membrane in activated HUVECs (×119 000). PM, plasma membrane. Panels E-G: PDI, 5-nm gold particles; SERCA2b, 15-nm gold particles.
PDI was also detected in tubulovescicular structures of the endoplasmic reticulum and in small granules close to the cell membrane by immunoelectron microscopy on ultrathin cryosections of HUVECs (Figure 3D). No PDI was observed in Weibel-Palade bodies. In agreement with the immunofluorescence data, immunogold labeling of PDI was observed in small, moderately electron-dense granules of approximately 100-200 nm (Figure 3E). In addition, immunogold staining confirmed the association of PDI with the plasma membrane of activated endothelial cells (Figure 3G), while no plasma membrane–associated PDI was detected in resting cells (Figure 3F).
PDI is released from cultured endothelial cells upon laser injury and is required for fibrin generation
Thrombin stimulation of human endothelial cells results in cell activation through the PAR1 receptor. In addition, we have demonstrated that endothelial cells can be activated by laser stimulation.32 Cell activation was monitored by calcium mobilization in vitro in cultured HUVECs. After a single laser pulse, HUVECs loaded with Fluo-4, a fluorescent calcium-sensitive dye, showed rapid elevation of intracellular calcium (Figure 4A). Activation of a single endothelial cell within a confluent culture also initiated a calcium wave that spread to the surrounding cells. Immunoblots of the supernatant medium collected from HUVECs before and after laser activation showed PDI antigen reactivity in the medium after laser activation. However, no PDI was detected in medium from HUVECs before laser activation (Figure 4A). In the presence of ultracentrifuged plasma, laser activation of HUVECs results in fibrin generation (Figure 4B left panel and 4C), which is inhibited in the presence of a function-blocking PDI antibody, RL90 (Figure 4B right panel and 4C). These results indicate that PDI released from activated endothelial cells is required for fibrin generation. In addition, in vivo laser activation of endothelium in a live mouse loaded with the calcium-sensitive dye, Rhod-2, showed rapid calcium elevation (pseudocolored green) captured in a confocal intravital image (Figure 4D). This observation is akin to the spread of the calcium wave observed in cultured endothelial cells, where laser activation of a single cell causes the subsequent activation of surrounding cells. In addition, after laser injury and the calcium wave spreading over the endothelium in vivo, PDI antigen was detected at the site of laser injury using a noninhibitory polyclonal affinity purified anti-PDI antibody labeled with Alexa 647.18 The PDI antigen postlaser injury was seen to spread around the circumference of the vessel wall, whereas no PDI was detected before endothelial activation (Figure 4D).

Rapid calcium mobilization and PDI expression after laser injury in vitro and in vivo. Fluo-4-AM (3μM) was incubated with cultured HUVECs before laser injury and observed by fluorescence microscopy. (A) The images show a representative field of cells before and after a direct laser pulse to the point indicated by X. Cell imaging was initiated before laser injury to obtain a baseline image, and the laser was fired during this capture. Increased green signal in subsequent time-lapse images represents increased intracellular calcium monitored by Fluo-4 fluorescence, shown in representative images. The Western blot on the right depicts an immunoblot with polyclonal anti-PDI antibody to detect PDI in conditioned medium from unactivated (lane 2) or laser-activated (lane 3) HUVECs. Lane 1: 2 ng recombinant human PDI. (B) Representative images of fixed and immunostained cells that have been activated by laser injury in the presence of plasma and calcium with (right) or without (left) a function blocking PDI antibody RL90. The cells were fixed after laser activation and stained for fibrin (red). Fluorescein isothiocyanate–phalloidin (green) and DAPI (4,6 diamidino-2-phenylindole; blue). (C) Quantification of fibrin signal detected on cultured endothelial: lane 1, unactivated cells; lane 2, laser-activated cells; lane 3, laser-activated cells in the presence of the PDI-inhibitory antibody RL90; lane 4, laser-activated cells in the presence of an isotype control antibody. The P values between the different groups were obtained using the unpaired t test. (D) Representative images of rapid activation of arteriolar endothelium and PDI expression after laser injury in confocal intravital microscopy. Rhod-2 (6μM) and Alexa 647–labeled polyclonal anti-PDI antibody (0.3 μg/g body weight) were infused 5 minutes before the first injury. Site of injury is indicated by an X. Platelet aggregation was blocked with the GPIIbIIIa antagonist, eptifibatide. Vessel imaging was initiated before laser injury, and the laser was fired after one z-stack of 30 planes was obtained. Each z-stack of 30 images was 8.7 seconds. The grid size was 10 μm. Calcium elevation was monitored by excitation of Rhod-2 at 561 nm (pseudocolored green), while the PDI signal was observed simultaneously at 647 nm (pseudocolored red).
Rapid calcium mobilization and PDI expression after laser injury in vitro and in vivo. Fluo-4-AM (3μM) was incubated with cultured HUVECs before laser injury and observed by fluorescence microscopy. (A) The images show a representative field of cells before and after a direct laser pulse to the point indicated by X. Cell imaging was initiated before laser injury to obtain a baseline image, and the laser was fired during this capture. Increased green signal in subsequent time-lapse images represents increased intracellular calcium monitored by Fluo-4 fluorescence, shown in representative images. The Western blot on the right depicts an immunoblot with polyclonal anti-PDI antibody to detect PDI in conditioned medium from unactivated (lane 2) or laser-activated (lane 3) HUVECs. Lane 1: 2 ng recombinant human PDI. (B) Representative images of fixed and immunostained cells that have been activated by laser injury in the presence of plasma and calcium with (right) or without (left) a function blocking PDI antibody RL90. The cells were fixed after laser activation and stained for fibrin (red). Fluorescein isothiocyanate–phalloidin (green) and DAPI (4,6 diamidino-2-phenylindole; blue). (C) Quantification of fibrin signal detected on cultured endothelial: lane 1, unactivated cells; lane 2, laser-activated cells; lane 3, laser-activated cells in the presence of the PDI-inhibitory antibody RL90; lane 4, laser-activated cells in the presence of an isotype control antibody. The P values between the different groups were obtained using the unpaired t test. (D) Representative images of rapid activation of arteriolar endothelium and PDI expression after laser injury in confocal intravital microscopy. Rhod-2 (6μM) and Alexa 647–labeled polyclonal anti-PDI antibody (0.3 μg/g body weight) were infused 5 minutes before the first injury. Site of injury is indicated by an X. Platelet aggregation was blocked with the GPIIbIIIa antagonist, eptifibatide. Vessel imaging was initiated before laser injury, and the laser was fired after one z-stack of 30 planes was obtained. Each z-stack of 30 images was 8.7 seconds. The grid size was 10 μm. Calcium elevation was monitored by excitation of Rhod-2 at 561 nm (pseudocolored green), while the PDI signal was observed simultaneously at 647 nm (pseudocolored red).
PDI is released from endothelial cells upon cell injury in vivo
PDI is expressed at the site of a developing thrombus in the laser-injury model, and platelets are one potential source of the PDI.18 The presence of an alternative source of PDI at sites of laser injury was suggested by the kinetics of PDI appearance before platelet accumulation during thrombus formation, and by the observation that fibrin generation was inhibited in the presence of a PDI-blocking antibody in PAR4−/− mice. In the latter experiment, fibrin generation, observed even in the absence of a significant platelet thrombus, was inhibited in the presence of inhibitory anti-PDI antibodies.18 The presence of PDI in plasma has been controversial.20,21 We examined both human and mouse plasma by immuoblotting for the presence of PDI. Comparison of the density units of immunoblots of purified recombinant human PDI from 0.02 to 1.75 pmol to the density units of PDI in human or mouse plasma demonstrates that 20 μL of these plasmas contain less than 0.02 pmol of PDI (supplemental Figure 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). To determine whether endothelial cells are an additional source of PDI, we compared PDI expression after laser-induced injury in mice in the presence and absence of eptifibatide, an antithrombotic agent that reversibly inhibits platelet aggregation by preventing the binding of fibrinogen to its receptor, αIIbβ3. PDI was visualized using a polyclonal affinity-isolated anti-PDI antibody directly labeled with Alexa Fluor 488. As previously described, at the concentrations used for imaging, this antibody does not inhibit PDI activity or perturb thrombus formation.18 Purified anti-CD41 Fab fragments were used to visualize platelet thrombus formation.33 Seven or 8 thrombi were induced in a wild-type mouse, and PDI expression and platelet accumulation were quantified. Subsequently, the mouse was treated with eptifibatide and additional thrombi were induced to determine the effect of inhibition of platelet thrombus formation on PDI expression.
Representative images of platelet thrombus formation and PDI expression after laser-induced injury before (left panels) and after (right panels) eptifibatide infusion are shown in Figure 5A. The median integrated platelet fluorescence for 30 thrombi generated in 7 mice before eptifibatide infusion indicates that platelet accumulation began at approximately 40 seconds after laser injury, and maximum platelet accumulation occurred at approximately 90-100 seconds (Figure 5B). The median integrated fluorescence associated with PDI for these same thrombi suggests that PDI expression preceded platelet accumulation, beginning almost immediately after laser injury and becoming maximal at approximately 25-30 seconds (Figure 5C). No platelet-associated fluorescence was observed in response to laser injury after eptifibatide infusion, as indicated by the median integrated fluorescence associated with platelet accumulation at the site of 30 laser injuries in 7 mice (Figure 5B). The median integrated fluorescence intensity for PDI at 30 laser injury sites in 7 mice generated after eptifibatide infusion followed similar kinetics to those in untreated mice for approximately 25 seconds. Median PDI fluorescence intensities at 15 seconds postinjury from 30 injuries, each in the presence and absence of eptifibatide, were not significantly different. There was a significant difference in median fluorescent intensities at 80 seconds after injury, with a value of P < .001, based on the 2-tailed Wilcoxon ranked-sum test. Similar results were observed after the infusion of R300 platelet-depleting antibody directed against mouse GP1bα (supplemental Figure 2). In addition, with inhibition of cell activation using a prostacyclin analog (sodium beraprost) in vivo, no PDI-associated fluorescence was detected (Figure 5D curve 2), compared with untreated animals (Figure 5D curve 1). These data indicate that both endothelial cells and platelets contribute to PDI accumulation at sites of laser-induced thrombus formation.

Comparison of PDI expression and platelet accumulation during thrombus formation. Rabbit polyclonal anti-PDI antibody conjugated to Alexa Fluor 488 (0.3 μg/g body weight) and Fab fragments of anti–CD-41 antibody conjugated to Alexa Fluor 647 (0.2 μg/g body weight) were infused into the mouse 5 minutes before arteriolar injury. In certain conditions, eptifibatide (10 μg/g body weight) was infused before injury and reinfused every 20 minutes for subsequent thrombi. (A). Representative binarized images of the appearance of fluorescence signals associated with PDI (green) and platelets (red) over 180 seconds after laser-induced vessel-wall injury in a wild-type (WT) mouse (left panels) or a wild-type mouse treated with eptifibatide. (B) Median integrated platelet fluorescence. Median fluorescence is presented vs. time after vessel-wall injury. Curve 1, WT mice; curve 2, WT mice treated with eptifibatide. (C-D) Median integrated PDI fluorescence detected by rabbit affinity-purified anti-PDI antibody. (D) Wild-type mouse treated with sodium beraprost (30 μg/kg body weight). Curve 1, WT mice; curve 2, WT mice treated with sodium beraprost.
Comparison of PDI expression and platelet accumulation during thrombus formation. Rabbit polyclonal anti-PDI antibody conjugated to Alexa Fluor 488 (0.3 μg/g body weight) and Fab fragments of anti–CD-41 antibody conjugated to Alexa Fluor 647 (0.2 μg/g body weight) were infused into the mouse 5 minutes before arteriolar injury. In certain conditions, eptifibatide (10 μg/g body weight) was infused before injury and reinfused every 20 minutes for subsequent thrombi. (A). Representative binarized images of the appearance of fluorescence signals associated with PDI (green) and platelets (red) over 180 seconds after laser-induced vessel-wall injury in a wild-type (WT) mouse (left panels) or a wild-type mouse treated with eptifibatide. (B) Median integrated platelet fluorescence. Median fluorescence is presented vs. time after vessel-wall injury. Curve 1, WT mice; curve 2, WT mice treated with eptifibatide. (C-D) Median integrated PDI fluorescence detected by rabbit affinity-purified anti-PDI antibody. (D) Wild-type mouse treated with sodium beraprost (30 μg/kg body weight). Curve 1, WT mice; curve 2, WT mice treated with sodium beraprost.
Endothelial cell–derived PDI is sufficient for maximal fibrin generation during laser-induced thrombosis
Inhibitory antibodies to PDI and the thiol isomerase inhibitor, bacitracin, block platelet thrombus formation and fibrin generation.18 To determine whether PDI secreted from endothelial cells alone is sufficient to support fibrin formation, we explored fibrin generation in eptifibatide-treated mice. A mouse anti-human fibrin IIβ-chain monoclonal antibody specific for human fibrin and crossreactive with mouse fibrin was directly labeled with Alexa Fluor 647 and used to monitor fibrin generation. Fibrin deposition at sites of laser injury after eptifibatide infusion was equivalent to that observed in the absence of the drug, indicating that the PDI released from endothelial cells after laser injury is sufficient to support maximal fibrin generation (Figure 6A,C). Infusion of both eptifibatide and the inhibitory anti-PDI monoclonal antibody resulted in an almost complete inhibition of fibrin deposition (Figure 6A,C). PDI expression at sites of laser injury was also decreased in the presence of RL90 (Figure 6B).

Inhibition of fibrin formation with a function-blocking PDI antibody is platelet independent. Rabbit polyclonal anti-PDI antibody conjugated to Alexa Fluor 488 (0.3 μg/g body weight) and fibrin-specific mouse anti–human fibrin II β-chain monoclonal antibody conjugated to Alexa Fluor 647 (0.5 μg/g body weight) were infused into the mouse 5 minutes before arteriolar injury. (A) Representative binarized images of the appearance of fluorescence associated with PDI (green) or fibrin (red) are shown over 180 seconds after laser-induced vessel-wall injury in a wild-type mouse (left panel), a wild-type mouse treated with eptifibatide (10 μg/g body weight; middle panel), or a wild-type mouse treated with eptifibatide (10 μg/g body weight) and a function blocking anti-PDI antibody, RL90, (2 μg/g body weight; right panel). Inhibitory monoclonal anti-PDI antibody RL90 and/or eptifibatide were infused into the circulation 5 minutes before injury. Data were collected from the same mouse pre- and postinfusion of eptifibatide and/or RL90. (B) Median integrated PDI fluorescence intensity or (C) median integrated fibrin fluorescence intensity for thrombi formed before (curve 1) or after the infusion of eptifibatide (curve 2) or after the infusion of eptifibatide in the presence of RL90 (curve 3). Median fluorescence is presented versus time after vessel wall injury.
Inhibition of fibrin formation with a function-blocking PDI antibody is platelet independent. Rabbit polyclonal anti-PDI antibody conjugated to Alexa Fluor 488 (0.3 μg/g body weight) and fibrin-specific mouse anti–human fibrin II β-chain monoclonal antibody conjugated to Alexa Fluor 647 (0.5 μg/g body weight) were infused into the mouse 5 minutes before arteriolar injury. (A) Representative binarized images of the appearance of fluorescence associated with PDI (green) or fibrin (red) are shown over 180 seconds after laser-induced vessel-wall injury in a wild-type mouse (left panel), a wild-type mouse treated with eptifibatide (10 μg/g body weight; middle panel), or a wild-type mouse treated with eptifibatide (10 μg/g body weight) and a function blocking anti-PDI antibody, RL90, (2 μg/g body weight; right panel). Inhibitory monoclonal anti-PDI antibody RL90 and/or eptifibatide were infused into the circulation 5 minutes before injury. Data were collected from the same mouse pre- and postinfusion of eptifibatide and/or RL90. (B) Median integrated PDI fluorescence intensity or (C) median integrated fibrin fluorescence intensity for thrombi formed before (curve 1) or after the infusion of eptifibatide (curve 2) or after the infusion of eptifibatide in the presence of RL90 (curve 3). Median fluorescence is presented versus time after vessel wall injury.
Discussion
We have determined that PDI is rapidly released from endothelial cells both in vitro and in vivo and contributes to the initiation of fibrin formation on activated endothelial cells in contact with plasma as well as in our laser injury thrombosis model. PDI is detected at the site of vessel wall injury in vivo in the absence of any platelet accumulation. Normal fibrin generation is observed even in the presence of an inhibitor of platelet aggregation, suggesting that PDI released from endothelial cells is critical for thrombus formation after laser-induced injury.
Because PDI antigen is not detected in plasma, any PDI observed at sites of laser injury in our thrombosis model is released from cells activated at the site. Catalytically active PDI can be released from activated platelets.20 However, the kinetics of PDI accumulation at sites of laser injury in the presence and the absence of eptifibatide suggest that PDI observed shortly after the generation of the laser-induced injury and before significant numbers of platelets accumulate is derived from endothelial cells. Platelets contribute to the more sustained PDI accumulation observed when platelet thrombus formation occurs. While lymphocytes have been shown to express PDI on the cell surface,34 leukocyte interaction with a growing thrombus does not begin until at least 2-3 minutes after laser injury, well after PDI appears.35 In addition, PDI levels and kinetics of appearance in PSGL-1 knock-out mice are comparable to wild-type mice, suggesting that PDI is released at the site of injury even in the absence of leukocyte-endothelium interactions (R.F., B.F., B.C.F., unpublished results, September 9, 2009). Thus, it is unlikely that leukocytes contributed to the PDI pool during our thrombosis experiments. Besides the vascular sources, PDI is also present in extravascular medial and adventitial layers of the vessel wall. However, our laser-injury model involved the activation of the intact endothelium without disruption of the vessel wall, ruling out any contribution of extravascular cells in releasing PDI at the site of injury.
Despite the absence of platelet accumulation at the site of laser-induced injury in the presence of eptifibatide, no differences in the kinetics of fibrin deposition or the quantity of fibrin accumulation were observed in the presence or absence of the drug. Fibrin accumulation is dependent on PDI, as the presence of both eptifibatide and the function blocking anti-PDI antibody RL90 dramatically reduces fibrin generation. These results are consistent with our previous observations that fibrin accumulation is normal in Par4−/− mice, even in the absence of significant platelet accumulation, and that the presence of the function blocking anti-PDI antibody RL90 inhibits fibrin formation in Par4−/− mice.18,22 Our current results emphasize the importance of PDI released from endothelial cells during laser-induced thrombus formation for fibrin formation independent of platelets.
Inhibition of fibrin formation, as a consequence of PDI inhibition both in cultured endothelial cells and in the in vivo mouse model, strongly implicates a role for PDI in thrombin generation. Furthermore, the role for PDI in thrombin formation is independent of platelets, as inhibition of PDI inhibits fibrin generation in the absence of platelet thrombus formation. Tissue factor, required for the initiation of thrombin generation in vivo, was present at sites of laser injury in our thrombosis model33 The thiol-isomerase activity of PDI has been implicated in regulating the activity of tissue factor.11,12 However, this mechanism of control of tissue factor activity remains controversial.13,36 Alternatively, PDI may regulate tissue factor activity by some mechanism unrelated to its oxidoreductive catalytic activity, or PDI may regulate the pathway to thrombin generation through mechanisms unrelated to platelet-associated molecules or tissue factor.
Activated platelets have generally been thought to provide the membrane surfaces on which the enzyme-cofactor complexes of blood coagulation assemble to support the catalytic reactions of these complexes. It is thus striking that we observed equivalent amounts of fibrin generated after laser-induced vessel wall injury in the presence or absence of a platelet thrombus. This result indicates that membrane surfaces other than those of the activated platelet may support thrombin generation, resulting in fibrin deposition. These other membrane surfaces could include endothelial cells and/or other membranes at the site of thrombus formation that have phosphatidylserine on the outer membrane surface. Cell-derived microparticles that express phosphatidylserine on their surface have previously been shown to accumulate at sites of thrombus formation after laser-induced injury.35 Platelet microparticles derived in vitro have been reported to bear detectable amounts of PDI.20,37 Hence, it is possible that both vessel wall endothelial cells and cell microparticles provide 2 of the necessary components, PDI and the membrane surface, to support fibrin deposition.
In summary, our data indicate that extracellular PDI accumulates at sites of laser injury, and both endothelial cells and platelets contribute to the available PDI. Inhibition of PDI had a dramatic effect on platelet thrombus formation and fibrin deposition in our laser injury model of thrombosis. PDI may represent a novel target for antithrombotic intervention, but the potential consequences of inhibition of extracellular PDI needs to be thoroughly evaluated in animal models to understand the possible risk-benefit ratio of such interventions.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Maria Ericsson for assistance with electron microscopy. We thank Glenn Merrill-Skoloff for expert technical assistance.
This work was supported by grants from the National Institutes of Health to B.F. and B.C.F. R.J. is a recipient of a fellowship from the American Heart Association.
National Institutes of Health
Authorship
Contribution: R.J., B.F., and B.C.F designed the research and wrote the paper; and R.J. performed the experiments and analyzed the data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Barbara C. Furie, Beth Israel Deaconess Medical Center, 330 Brookline Ave, E/CLS, RM 905, Boston, MA 02215; e-mail: bfurie1@bidmc.harvard.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal