

Abstract
Preclinical studies of BCR-ABL mutation sensitivity to nilotinib or dasatinib suggested that the majority would be sensitive. Correspondingly, the initial clinical trials demonstrated similar response rates for CML patients after imatinib failure, irrespective of the mutation status. However, on closer examination, clinical evidence now indicates that some mutations are less sensitive to nilotinib (Y253H, E255K/V, and F359V/C) or dasatinib (F317L and V299L). T315I is insensitive to both. Novel mutations (F317I/V/C and T315A) are less sensitive/insensitive to dasatinib. We refer to these collectively as second-generation inhibitor (SGI) clinically relevant mutations. By in vitro analysis, other mutations confer a degree of insensitivity; however, clinical evidence is currently insufficient to define them as SGI clinically relevant. Here we examine the mutations that are clearly SGI clinically relevant, those with minimal impact on response, and those for which more data are needed. In our series of patients with mutations at imatinib cessation and/or at nilotinib or dasatinib commencement, 43% had SGI clinically relevant mutations, including 14% with T315I. The frequency of SGI clinically relevant mutations was dependent on the disease phase at imatinib failure. The clinical data suggest that a mutation will often be detectable after imatinib failure for which there is compelling clinical evidence that one SGI should be preferred.
Introduction
The outcome for patients with chronic myeloid leukemia (CML) who fail imatinib has improved since the availability of second-generation BCR-ABL kinase inhibitors (SGIs). The most common mechanisms of imatinib resistance are mutations within the BCR-ABL kinase domain and protein overexpression by gene amplification.1-12 Resistance is also associated with other genetic events, as indicated by the detection of cytogenetic abnormalities in the Philadelphia chromosome–positive (Ph+) clone in more than 50% of imatinib-resistant patients.13 It has been suggested that some BCR-ABL mutations play no causal role in resistance.14-16 However, approximately half of the patients who commence SGIs after imatinib therapy have detectable imatinib-resistant BCR-ABL mutations. Imatinib binds to the inactive conformation of BCR-ABL, leading to disruption of the adenosine triphosphate (ATP) binding site and blockade of the catalytic activity.17,18 BCR-ABL mutations that impair imatinib binding while still enabling ATP binding, or that alter the specific protein conformation required for imatinib binding, are selected in the presence of imatinib.19-21 In the absence of imatinib, these mutations do not confer a growth advantage.22
For patients commencing nilotinib or dasatinib after imatinib cessation, clinical trials have demonstrated similar responses for patients with or without mutations, except for T315I for which neither drug is active.23-31 This mutation demonstrates cross-resistance to imatinib, nilotinib, and dasatinib.32-34 However, a closer examination of responses to SGI therapy for individual mutations has identified a limited number, other than T315I, that are less sensitive to either nilotinib or dasatinib.35-37 Furthermore, in vitro studies have identified mutations that confer a degree of insensitivity38 or resistance.39
How well do the problematic mutations identified by in vitro studies correlate with those identified by clinical studies? Moreover, does the in vitro sensitivity of mutations provide a reliable indication of the probable response to SGIs? Undoubtedly, in vitro sensitivity of imatinib-resistant mutations can be a useful guide when considering an increased imatinib dose.40 Here we assess BCR-ABL mutations in the context of their impact on response after a change to SGI therapy by an examination of the available clinical data. The mutation status may contribute to therapeutic decisions after imatinib failure or indeed after failure of an SGI. We assess the frequency that mutations conferring a degree of clinical insensitivity to SGIs are detectable at the time of imatinib cessation. These are collectively referred to as SGI clinically relevant mutations. We also examine whether the disease phase influences their frequency. Last, we examine the occurrence of multiple mutations in imatinib-treated patients and the extent to which disease phase influences their detection.
BCR-ABL mutations in the era of SGIs: type still matters
Mutant sensitivity assessed by in vitro studies
Preclinical studies of nilotinib against 33 BCR-ABL mutants predicted that the inhibitor would have clinical activity in patients harboring these mutations, except for T315I.33,34,41 Similarly, among 19 imatinib-resistant mutants tested against dasatinib, T315I was the only clearly resistant mutation.32,33 The in vitro results were similar to the earlier in vitro studies of imatinib, in that mutants displayed various degrees of sensitivity.15,42 SGI sensitivity was assessed by various methods, including the degree of inhibition of BCR-ABL autophosphorylation or cell proliferation after transfection of mutants into Ba/F3 cells.32-34 Although the mutations conferred various degrees of sensitivity to the SGIs, all except T315I were predicted to be sensitive at clinically achievable doses. Resistance was predicted to be associated with the predominant emergence of T315I. However, the concentration of drug required to kill Ba/F3 cells transformed to interleukin-3 independence by BCR-ABL cannot be assumed to be representative of the concentration required to kill primary CML progenitor cells in vivo.
There is currently no uniform reporting format for in vitro mutant sensitivity to SGIs, and discrepancies in the reported sensitivity exist between studies. Sensitivity is described as an IC50 value38 or as the fold increase in IC50 relative to wild-type BCR-ABL, which is expressed as 1.39 Mutants are described variously as sensitive, intermediate sensitivity, and insensitive,38 or sensitive, moderately resistant, resistant, and highly resistant.39 T315I is consistently described as resistant/highly resistant, and responses are unexpected. However, what about other mutants classified as conferring intermediate sensitivity or as resistant/highly resistant to nilotinib or dasatinib, as summarized in Table 1? Does this indicate that responses are unexpected or may be inferior for patients harboring these mutations?
Order of mutation sensitivity to dasatinib and nilotinib as determined by in vitro cell proliferation assays. Shading of O'Hare: red, resistant; yellow, intermediate sensitivity; and green, sensitive. Shading of Redaelli: red, highly resistant; orange, resistant; yellow, moderately resistant; and green, sensitive. Bold type indicates mutations where there is a difference in the classification between the studies. *Although T315A is rated as resistant, it is rarely detected.
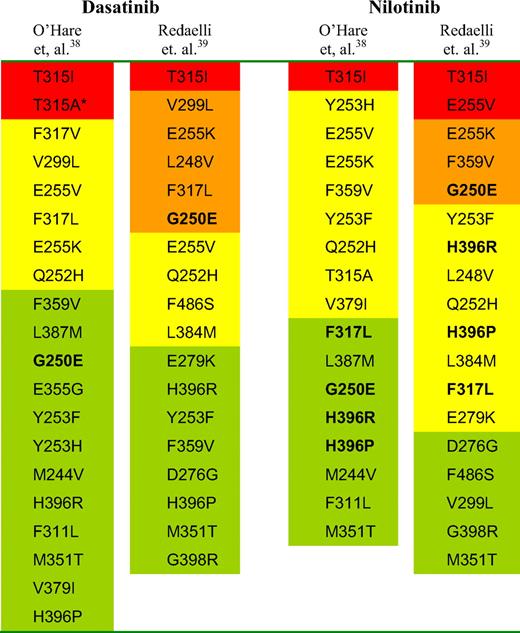
The reliability of in vitro sensitivity classifications is uncertain and requires further clinical validation. If validated, they may prove extremely useful to provide guidance when considering appropriate therapeutic intervention after inhibitor failure. However, interpreting the sensitivity for some mutants from in vitro studies is not straightforward. For example, the common imatinib-resistant mutation G250E is reported in one study as resistant to imatinib, nilotinib, dasatinib, and indeed, to another inhibitor bosutinib, which is under clinical trial as SGI therapy.39 However, G250E is described as sensitive to nilotinib and dasatinib in another assessment.38 F317L is classified as moderately resistant or resistant to the 4 inhibitors in one study39 and sensitive to nilotinib and insensitive to dasatinib in another.38 These discrepancies may be the result of methodologic differences or differences in the cutoff values for the classification of mutant sensitivity and may cause difficulty for interpretation.
Mutations identified from resistance screens
Resistance screens have identified a limited number of mutations that emerge in the presence of increasing doses of nilotinib and dasatinib,43-46 and these correspond, to a degree, to the mutant sensitivity determined in cell proliferation assays.38,39 Mutations at dasatinib contact residues appeared to be particularly relevant, including V299L. In 2 resistance screens, mutations at T315 and F317 accounted for 95% of all mutants recovered, including novel mutations F317V/I/S/C and T315A, which had not been reported in imatinib-treated patients.44,45 In one study, F317V and T315A were the most frequent to emerge (41% and 30%, respectively) and had 40- to 90-fold reduced dasatinib sensitivity compared with unmutated BCR-ABL.45 In accord with these results, T315A and F317V had the highest IC50 values, except for T315I, in the in vitro assessment of dasatinib by O'Hare et al.33
In 3 in vitro resistance screens of increasing doses of nilotinib, T315I emerged most frequently and represented 49% of all mutations recovered.43,44,46 However, the common imatinib-resistant mutation, Y253H, was also among the most frequent to emerge and had the highest nilotinib IC50 value in each of these studies, apart from T315I. E255K, Y253H, and T315I were the only mutations to emerge in all 3 screens, and E255V and Q252H emerged in 2 of 3 studies. All other mutations were confined to one of the screens. With the exception of T315I, all mutations were effectively suppressed by nilotinib concentrations of 2000nM, which falls within the peak-trough plasma levels (3600-1700nM) measured in patients treated with 400 mg nilotinib twice daily.24
Over the past few years, clinical studies have identified a limited number of mutations that may be relevant for response to nilotinib and dasatinib as second- or third-line inhibitor therapy35,36,47,48 and are implicated in resistance.47-51 In one report, a new mutation was acquired in 83% of patients who relapsed after a response.48 The available clinical data present an opportunity to assess how effectively in vitro studies predict the SGI clinically relevant mutations and their validity for determining appropriate therapy after imatinib failure.
Mutation sensitivity assessed by clinical studies of dasatinib
The majority of imatinib-resistant mutations remain sensitive to dasatinib.23,26-29,31 However, consistent with the dasatinib resistance screens and cell proliferation assays, clinical reports confirmed T315I/A, F317L/I/V/C, and V299L as relevant for decreased clinical efficacy, either as preexisting or as emerging mutations.37,48-51
One of the most frequent mutations to emerge with clinical dasatinib resistance was V299L.48,50 This mutation was reported very rarely in imatinib-treated patients.8,52 V299L and F317L were also preferentially associated with dasatinib failure in a study of mutation dynamics after sequential inhibitor therapy.51 Interestingly, V299L was only detected at a frequency of 1% in resistance screens44,45 and only at lower dasatinib concentrations.44
The largest analysis of clinical response to dasatinib after imatinib failure to date involved 1043 CML patients treated in chronic phase (CP).37 The presence of T315I or F317L at the time of commencing dasatinib was associated with the least favorable responses. Furthermore, the most frequently detected new mutations were T315I, F317L, and V299L. A conclusion of the study was that alternative treatment options should be considered for patients with these mutations.37 Consistent with these findings, other studies reported low response rates for patients with F317L.47,48 In one study, 8 of 16 dasatinib-treated patients after imatinib failure acquired F317L, and this mutation was deemed dasatinib-resistant but sensitive to other inhibitors.47
Mutant sensitivity assessed by clinical studies of nilotinib
Clinical studies have demonstrated that the majority of imatinib-resistant mutations remain sensitive to nilotinib.24,25,30 Nevertheless, several mutations are less sensitive, which influences the response.36 The mutations that emerged in the nilotinib resistance screens have corresponded, to a degree, with the clinical findings. In an evaluation of 281 CP patients in the nilotinib phase 2 registration study, those with T315I, Y253H, E255K/V, and F359V/C (n = 31) at nilotinib commencement had the least favorable responses. These mutations had the highest IC50 values in cell proliferation assays as assessed by Weisberg et al (> 150nM).34,41 No patient with these mutations achieved a complete cytogenetic response (CCyR) by 12 months, 6 (19%) achieved a major cytogenetic response, and 10 (32%) a complete hematologic response (CHR).36 In contrast, 32 of 74 patients (43%) with any other mutation and 35 of 87 (40%) with imatinib resistance but no mutation achieved CCyR. These mutations were also among the most common new mutations during nilotinib therapy and were associated with a higher risk of progression. In another study, 13 of 14 patients who relapsed with new mutations on nilotinib as second- or third-line inhibitor therapy had one of these mutations.48
The poor response associated with Y253H, E255K/V, and F359V/C when present at the time of commencing nilotinib was confirmed in patients treated in accelerated phase (AP) CML.53 Of 17 of 87 AP patients with these mutations, only 24% achieved a CHR. In contrast, 55% without mutations and 58% with other mutations (excluding T315I) achieved a CHR. From the nilotinib clinical response data, the suggestion was that therapies other than nilotinib should be considered for patients with these mutations.36,53 Consistent with these findings, the most frequent mutations detected in patients with nilotinib failure in the study of Cortes et al51 were at residues 253, 255, 359, and 311. A mutation at residue 311 was also detected in a nilotinib resistance screen.43
Which mutations are relevant for response and resistance to nilotinib and dasatinib from clinical studies?
The current clinical data suggest the SGI clinically relevant mutations are T315I for both inhibitors: F317L/I/C/V, V299L, and T315A for dasatinib and Y253H, E255K/V, and F359V/C for nilotinib. Sensitive mutation detection of these specific mutations to aid therapeutic choices may be beneficial. Several techniques moderately improve the sensitivity to 1.5% to 10%, including pyrosequencing,16,51 ligation-dependent competitive polymerase chain reaction (PCR),54 and SEQUENOM MassARRAY.55 Highly sensitive techniques have a detection limit from 0.0003% to 0.1%, including mutation-specific PCR based on the Taqman platform,56 PCR–restriction fragment length polymorphism,57 polymerase colony assay,58 allele-specific PCR,59 and a nanofluidic platform.60 Whether highly sensitive detection of SGI clinically relevant mutations before SGI therapy will always correlate with their clonal expansion and resistance is unknown. This was not always the case using highly sensitive mutation detection before imatinib therapy.59
Do the SGI clinically relevant mutations correspond to the in vitro data?
Based on current clinical information, the answer to this question is yes, to a degree. Why are some mutations clinically relevant for SGIs and not others that either emerged more frequently in in vitro resistance screens and/or those with greater in vitro insensitivity? In the case of dasatinib, identification of clinically relevant mutations at residues T315 and F317 is consistent with their emergence in resistance screens. However, despite the high frequency of F317V and T315A in a dasatinib resistance screen and their significantly reduced sensitivity to dasatinib compared with F317L,45 F317V was not detected in any patient and T315A in only 2 patients in the initial reports.49,50 Could this be related to reduced oncogenicity of these mutations? Severely attenuated transforming activity of T315A was demonstrated relative to wild-type BCR-ABL.61 However, F317L was only marginally more transforming than T315A. Furthermore, reduced transforming activity does not appear to be related to frequency of detection of imatinib-resistant mutations: M351T displays reduced transforming activity61,62 yet is among the most commonly detected imatinib-resistant mutations.63
The sensitivity rankings by cell proliferation assays consistently suggest that the P-loop mutations Q252H and E255K/V may be relevant for dasatinib (Table 1).38,39 In dasatinib-treated CP patients, the CCyR rates for patients with these mutations ranged from 17% to 38%.37 These mutations have rarely been associated with clinical dasatinib resistance or as new mutations during dasatinib therapy.37,48-51 However, E255K and Q252H were among the mutations recovered in in vitro dasatinib resistance screens but were the only noncontact residues.44,45 The BCR-ABL crystal structure in complex with dasatinib suggested that interactions between the P-loop and dasatinib were less critical for binding.64 Manley et al65 proposed that it is doubtful mutations of Q252 and E255 could cause a change in the structure of the P-loop to interfere with dasatinib binding, without also disturbing binding of ATP, which is critical for BCR-ABL reactivation. Clearly, additional clinical information is required before the significance for dasatinib response of E255K/V and Q252H is elucidated.
Nilotinib in vitro sensitivity classifications38,39 correlate very closely with clinical data (Table 1). T315I, Y253H, E255K/V, and F359V have the highest IC50 values (F359C was not tested). The major inconsistency is for the classification of G250E (sensitive38 /resistant39 ). The IC50 reported by Weisberg et al41 for G250E was 145nM, which is close to the cutoff value of 150nM used to define mutations less sensitive to nilotinib in the clinical evaluation of CP patients.36 There were only 5 CP patients with this mutation at nilotinib start, and 3 (60%) achieved a CCyR.36 G250E was among the most common mutations to emerge with nilotinib.36 However, it was not among the most common mutations associated with progression. Mutations that are less sensitive, but still responsive, to nilotinib or dasatinib may mistakenly appear to be newly acquired as more sensitive alleles disappear more rapidly. G250E did not emerge in another study in which 13 patients with nilotinib resistance acquired new mutations.48 Inconsistency is also apparent for in vitro sensitivity classification of G250E for dasatinib (sensitive38 /resistant39 ). This mutation was the most commonly detected in CP patients at the start of dasatinib, and 20 of 60 (33%) achieved a CCyR.37 G250E did not emerge in the resistance screens of dasatinib.44,45 There is currently no strong clinical evidence to suggest that the presence of G250E would influence the response to nilotinib or dasatinib. Q252H and Y253F are consistently classified as moderately insensitive38 or resistant39 to nilotinib by in vitro assessment, and Q252H emerged in the nilotinib resistance screens.43,46 Further clinical data are required for adequate assessment of their response to nilotinib.
Validation of in vitro sensitivity of different mutations was recently demonstrated.35 In vitro sensitivity was predictive of response and long-term outcome for patients treated with nilotinib or dasatinib. Mutations for which there was a discrepancy in reported sensitivity among in vitro studies were classified according to the highest IC50 to the corresponding inhibitor. However, G250E was classified as a sensitive mutation to both inhibitors despite the discrepancy in the in vitro sensitivity classifications.38,39 In Figure 1, CCyR rates of CP patients with various mutations at the start of dasatinib therapy in the large clinical study of Müller et al37 are plotted according to in vitro sensitivity classifications. CCyR rates were only partially predicted by in vitro sensitivity.

Correlation of CCyR rates and the in vitro mutation sensitivity rankings for CP patients treated with dasatinib. The CCyR rates of patients with various imatinib-resistant mutations treated in trials of dasatinib37 were plotted according to the in vitro sensitivity classification of (A) Redaelli et al39 (n = 286 patients) and (B) O'Hare et al38 (n = 335 patients). The mutations are ranked in order of in vitro sensitivity (highest to lowest from the left). The number of patients with each mutation at dasatinib start ranged from 5 for L387M to 60 for G250E. The cumulative CCyR rate for all patients with mutations in the study was 43%. CCyR rates between 30% and 40% were achieved for patients with sensitivity classifications of (A) resistant (orange), moderately resistant (yellow), and sensitive (green), and (B) intermediate sensitivity (yellow) and sensitive (green). Only patients with T315I or F317L, which are classed as SGI clinically relevant, had CCyR rates less than 10%. This suggests that the in vitro sensitivity was only partially predictive of the clinical response to dasatinib.
Correlation of CCyR rates and the in vitro mutation sensitivity rankings for CP patients treated with dasatinib. The CCyR rates of patients with various imatinib-resistant mutations treated in trials of dasatinib37 were plotted according to the in vitro sensitivity classification of (A) Redaelli et al39 (n = 286 patients) and (B) O'Hare et al38 (n = 335 patients). The mutations are ranked in order of in vitro sensitivity (highest to lowest from the left). The number of patients with each mutation at dasatinib start ranged from 5 for L387M to 60 for G250E. The cumulative CCyR rate for all patients with mutations in the study was 43%. CCyR rates between 30% and 40% were achieved for patients with sensitivity classifications of (A) resistant (orange), moderately resistant (yellow), and sensitive (green), and (B) intermediate sensitivity (yellow) and sensitive (green). Only patients with T315I or F317L, which are classed as SGI clinically relevant, had CCyR rates less than 10%. This suggests that the in vitro sensitivity was only partially predictive of the clinical response to dasatinib.
There may be rare imatinib-resistant mutations not included in in vitro studies that could also be less sensitive to SGIs, such as F359I. This mutation emerged in one nilotinib resistance screen.43 Furthermore, rapid progression was observed in a nilotinib-treated patient harboring F359I at commencement of nilotinib.66 The IC50 value of this mutation is unknown.
From the available studies, we now have a clearer understanding of the BCR-ABL mutations for which there is compelling clinical evidence that response could be compromised by treatment with one and/or another of the SGIs if present after imatinib failure. These are T315I, F317L, V299L, Y253H, E255K/V, and F359V/C, the finding of which would influence the therapeutic decision. These mutations are classified in Table 2 as either class D (no role for SGI therapy) or class C (compelling clinical evidence to recommend an alternative inhibitor). At this stage, the presence of other mutations should have no impact on clinical decisions. Nevertheless, there are mutations where further clinical evidence may reveal relevance for an inhibitor (class B, Table 2). However, additional clinical assessment is required before an alternative inhibitor would be recommended for class B mutations. Table 2 lists the frequency of mutations at our institution, whereas the classifications are based on published literature. Of course, mutation status is only one factor that needs to be considered when selecting SGIs, which include issues of tolerance and the disease phase.
How frequently will the mutation status be an issue when considering therapeutic options after imatinib failure?
We have performed BCR-ABL mutation analysis at our institution for imatinib-treated patients since 2001, which allows an assessment of the frequency of SGI clinically relevant mutations at a single institution. Mutations were detected in 386 patients using direct sequencing5,67,68 at the time of imatinib cessation (n = 159) and/or at commencement of nilotinib or dasatinib after imatinib failure (n = 227). The assay has a mutation sensitivity of 10% to 20%, and the approximate percentage of mutant nucleotide relative to unmutated nucleotide is calculated either by the mutation detection software or, for low-level mutations, by differences in peak height on the sequencing chromatogram. The analysis was conducted in accordance with the Declaration of Helsinki, and informed consent was obtained.
Range of mutations
In total, 503 mutations were detected, which were composed of 55 different nucleotide exchanges at 34 residues. Table 2 lists the 20 mutations detected at frequencies of more than 1% of all mutations. These 20 accounted for 88% of all mutations. T315I was the most frequent (14% of all patients with mutations), and 83% of the mutations had known nilotinib and dasatinib IC50 values calculated by in vitro cell proliferation.38,39 A collation of more than 800 mutations derived from the literature also found that T315I was most frequently detected.63 In that collation, the 7 most common mutations accounted for 66% of all mutations and occurred at residues T315, Y253, E255, M351, G250, F359, and H396. In our cohort, these mutations accounted for 60%, indicating reasonable agreement between the 2 collations.
Most frequent mutations detected at a single institution, which accounted for 88% of all mutations
| Mutation | No. detected | Percentage of patients with mutations (n = 386) | Percentage of all mutations (n = 503) | Mutation class for therapeutic decision* | |
|---|---|---|---|---|---|
| Nilotinib | Dasatinib | ||||
| T315I | 53 | 13.7 | 10.6 | D | D |
| M351T | 47 | 12.2 | 9.4 | A | A |
| G250E | 46 | 11.9 | 9.2 | A | A |
| F359V | 35 | 9.1 | 7.0 | C | A |
| M244V | 33 | 8.5 | 6.6 | A | A |
| Y253H | 32 | 8.3 | 6.4 | C | A |
| E255K | 27 | 7.0 | 5.4 | C | B |
| H396R | 26 | 6.7 | 5.2 | A | A |
| F317L | 22 | 5.7 | 4.4 | A | C |
| E355G | 16 | 4.1 | 3.2 | A | A |
| Q252H | 15 | 3.9 | 3.0 | B | B |
| E255V | 14 | 3.6 | 2.8 | C | B |
| E459K | 14 | 3.6 | 2.8 | A | A |
| F486S | 13 | 3.4 | 2.6 | A | A |
| L248V | 10 | 2.6 | 2.0 | A | A |
| D276G | 10 | 2.6 | 2.0 | A | A |
| E279K | 10 | 2.6 | 2.0 | A | A |
| Y253F | 6 | 1.6 | 1.2 | B | A |
| F359C | 6 | 1.6 | 1.2 | C | A |
| F359I | 6 | 1.6 | 1.2 | B | A |
| Mutation | No. detected | Percentage of patients with mutations (n = 386) | Percentage of all mutations (n = 503) | Mutation class for therapeutic decision* | |
|---|---|---|---|---|---|
| Nilotinib | Dasatinib | ||||
| T315I | 53 | 13.7 | 10.6 | D | D |
| M351T | 47 | 12.2 | 9.4 | A | A |
| G250E | 46 | 11.9 | 9.2 | A | A |
| F359V | 35 | 9.1 | 7.0 | C | A |
| M244V | 33 | 8.5 | 6.6 | A | A |
| Y253H | 32 | 8.3 | 6.4 | C | A |
| E255K | 27 | 7.0 | 5.4 | C | B |
| H396R | 26 | 6.7 | 5.2 | A | A |
| F317L | 22 | 5.7 | 4.4 | A | C |
| E355G | 16 | 4.1 | 3.2 | A | A |
| Q252H | 15 | 3.9 | 3.0 | B | B |
| E255V | 14 | 3.6 | 2.8 | C | B |
| E459K | 14 | 3.6 | 2.8 | A | A |
| F486S | 13 | 3.4 | 2.6 | A | A |
| L248V | 10 | 2.6 | 2.0 | A | A |
| D276G | 10 | 2.6 | 2.0 | A | A |
| E279K | 10 | 2.6 | 2.0 | A | A |
| Y253F | 6 | 1.6 | 1.2 | B | A |
| F359C | 6 | 1.6 | 1.2 | C | A |
| F359I | 6 | 1.6 | 1.2 | B | A |
Class A indicates currently no compelling clinical evidence to suggest that the mutation would not respond to the inhibitor. Class B, In vitro assessment consistently indicates that the mutation may confer intermediate insensitivity38/resistance39 to the inhibitor, or clinical evidence may be suggestive of reduced sensitivity. At this stage, the presence of these mutations should have no impact on clinical decisions and additional clinical assessment is required before an alternative inhibitor would be recommended. Class C, Compelling clinical evidence to recommend an alternative inhibitor; V299L, which is very rarely detected in imatinib-treated patients, is a dasatinib class C mutation. Class D, No role for SGI therapy.
Difference in the types of mutations according to the disease phase
The disease phase at imatinib cessation and/or commencement of nilotinib or dasatinib after imatinib failure was known for 311 patients (81%): lymphoid blast crisis/Ph+ acute lymphoblastic leukemia (LBC/Ph+ ALL), n = 52, myeloid blast crisis (MBC), n = 50, AP, n = 76 and CP, n = 133. This disease phase classification is different from other reports, which classified patients according to the phase at imatinib commencement.5-8 There were noticeable differences in individual mutation frequency among the disease phases. Table 3 lists the mutations detected in more than 5% of patients within their disease phase. The mutations within each phase were partially overlapping, and only 4 were shared among the sets of common mutations: T315I, Y253H, G250E, and M244V.
Percentage of patients with the most common individual mutations in each of the disease phases
| Disease phase/mutation | Percentage |
|---|---|
| LBC/Ph+ ALL (n = 52) | |
| T315I | 21.2 |
| E255K | 19.2 |
| Y253H | 15.4 |
| G250E | 13.5 |
| E255V | 7.7 |
| F317L | 7.7 |
| M244V | 5.8 |
| F359V | 5.8 |
| E459K | 5.8 |
| MBC (n = 50) | |
| T315I | 16.0 |
| G250E | 14.0 |
| F486S | 12.0 |
| M244V | 10.0 |
| Q252H | 10.0 |
| M351T | 10.0 |
| H396R | 8.0 |
| L248V | 6.0 |
| Y253H | 6.0 |
| AP (n = 76) | |
| F359V | 14.5 |
| T315I | 13.2 |
| M351T | 13.2 |
| H396R | 10.5 |
| Y253H | 9.2 |
| M244V | 7.9 |
| G250E | 6.6 |
| E355G | 6.6 |
| Q252H | 5.3 |
| E255K | 5.3 |
| E279K | 5.3 |
| CP (n = 133) | |
| M351T | 16.5 |
| G250E | 12.0 |
| M244V | 11.3 |
| F359V | 9.8 |
| T315I | 7.5 |
| H396R | 7.5 |
| Y253H | 6.8 |
| F317L | 6.0 |
| E355G | 5.3 |
| Disease phase/mutation | Percentage |
|---|---|
| LBC/Ph+ ALL (n = 52) | |
| T315I | 21.2 |
| E255K | 19.2 |
| Y253H | 15.4 |
| G250E | 13.5 |
| E255V | 7.7 |
| F317L | 7.7 |
| M244V | 5.8 |
| F359V | 5.8 |
| E459K | 5.8 |
| MBC (n = 50) | |
| T315I | 16.0 |
| G250E | 14.0 |
| F486S | 12.0 |
| M244V | 10.0 |
| Q252H | 10.0 |
| M351T | 10.0 |
| H396R | 8.0 |
| L248V | 6.0 |
| Y253H | 6.0 |
| AP (n = 76) | |
| F359V | 14.5 |
| T315I | 13.2 |
| M351T | 13.2 |
| H396R | 10.5 |
| Y253H | 9.2 |
| M244V | 7.9 |
| G250E | 6.6 |
| E355G | 6.6 |
| Q252H | 5.3 |
| E255K | 5.3 |
| E279K | 5.3 |
| CP (n = 133) | |
| M351T | 16.5 |
| G250E | 12.0 |
| M244V | 11.3 |
| F359V | 9.8 |
| T315I | 7.5 |
| H396R | 7.5 |
| Y253H | 6.8 |
| F317L | 6.0 |
| E355G | 5.3 |
A total of 13% of patients had more than 1 of these mutations.
Because the mutation status of some patients may have been known when the clinician committed to SGI therapy, there is the possibility that our cohort is underrepresentative of T315I. This could be the case if clinicians pursued other therapeutic options for their patients with T315I. However, comparison of T315I frequency between samples that were only received after commitment to SGI therapy and those from patients where the mutation status was known at imatinib cessation indicates that the frequency is comparable, 13.2% and 14.5%, respectively.
Mutations with the most contrasting frequencies among the disease phases are highlighted in Figure 2. Even among patients with BC, there were stark differences according to phenotype. Q252H and F486S were associated with MBC (22%) compared with LBC/Ph+ ALL (3.8%). A higher frequency of Y253H and E255K/V was evident in LBC/Ph+ ALL (42%) compared with MBC (12%). This difference was not the result of mutation frequency differences among patients with Ph+ ALL compared with LBC. Of the 31 patients with Ph+ ALL, the frequency of Y253H and E255K/V was 45%, whereas in the 21 patients with LBC it was 38%. Some patients had more than one of these mutations. Consistent with our finding, E255K/V and Y253H were reported in 11 of 16 patients (69%) with LBC/Ph+ ALL in 2 small series.4,69 Furthermore, Y253, E255, T315, Q252, F486, and E459 were associated with advanced-phase disease in the compilation of more than 800 mutations, but the BC phenotype was not distinguished.63

Mutations with the greatest difference in frequency among the disease phases. T315I was most frequently detected in patients with LBC/Ph+ ALL, whereas M351T was most frequently detected in CP. Differences were observed according to the blast crisis phenotype. Y253H and E255K/V were associated with LBC/Ph+ ALL, whereas Q252H and F486S were associated with MBC.
Mutations with the greatest difference in frequency among the disease phases. T315I was most frequently detected in patients with LBC/Ph+ ALL, whereas M351T was most frequently detected in CP. Differences were observed according to the blast crisis phenotype. Y253H and E255K/V were associated with LBC/Ph+ ALL, whereas Q252H and F486S were associated with MBC.
It is possible that higher-dose imatinib, which is frequently used in advanced phases, will lead to a greater proportion of mutations that are highly resistant, such as T315I and P-loop mutations. This could account for the greater proportion of these mutations in advanced phases in our analysis. However, we classified the patients according to their disease phase at imatinib cessation and/or at commencement of SGI therapy, which was largely independent of the dose. Soverini et al9 classified 127 imatinib-treated patients with mutations according to the disease phase at first evidence of imatinib resistance, which is similar to our classification. Consistent with our findings, P-loop and T315I mutations were particularly frequent in BC and Ph+ ALL and often accompanied progression from CP to AP/BC. Furthermore, patients with P-loop or T315I mutations had a poorer survival or higher rates of progression to advanced phase than other mutations, irrespective of the disease phase at imatinib start.5-7,9,12 Therefore, the higher rate of detection of these mutations in patients who progressed to advanced phases in our study is highly consistent. However, there are some inconsistencies in our mutation frequency. In AP, some mutation frequencies were intermediate between CP and BC (Figure 2), which could be associated with transition between the phases. However, F359V was the most frequent mutation detected in AP but detected at a relatively low frequency in BC. The frequencies of individual mutations in specific disease phases should be considered with caution, and validation is required.
The differences evident in BC phenotype, if validated, raise several questions. Why were these particular mutations related to either a lymphoid or myeloid phenotype? Could the BCR-ABL mutant genotype drive the lineage phenotype in some cases or vice versa? A known genetic determinant of LBC is homozygous deletion of the p16INK4A/p19ARF gene locus that occurs in a substantial proportion of LBC patients, but not MBC.70-74 It was postulated that p16INK4A and/or p19ARF mutations could induce the selective expansion of B-cell progenitors by favoring their cell cycle entry, rather than myeloid progenitors.75
In the case of BCR-ABL mutations, in vitro transformation assays have demonstrated a gain or loss of function relative to unmutated BCR-ABL.61,62 Mutations can alter the substrates that bind to BCR-ABL and activate alternate signal transduction pathways that influence disease progression. Interestingly, E255K and Y253F mutations showed a pronounced increase of transformation potency in primary B-lymphoid progenitor cells.62 However, the transformation potency of these mutations was not significantly increased over unmutated BCR-ABL in the myeloid lineage. In our cohort, the frequency of E255K was markedly increased in LBC/Ph+ ALL (19.2%) compared with MBC (2.0%). However, Y253F was detected at a low frequency in all disease phases. The SRC kinases, LYN, HCK, and FGR, have also been implicated in the molecular pathogenesis of Ph+ ALL76 and are critical for transition of CML to LBC.77 Whether BCR-ABL mutations that are characteristic of BCR-ABL+ lymphoid leukemia in our cohort enhance the activation of SRC kinases and hence the transition to a lymphoid phenotype is unknown.
Frequency of mutations that would influence the therapeutic decision
In our cohort of patients with mutations, 166 of 386 (43%) had one or more SGI clinically relevant mutations. T315I was detected in 53 of 386 patients (14%). In 110 of 386 patients (28%), one or more of their mutations were clinically relevant for either nilotinib or dasatinib, but not to both. For these patients, there may be an advantage for one or other inhibitor. The remaining 3 of 386 patients (0.8%) had 2 mutations, one of which was clinically relevant for dasatinib and the other for nilotinib (neither was T315I).
Among the disease phases, there were significant differences in frequency of SGI clinically relevant mutations: 63% LBC/Ph+ ALL, 32% MBC, 49% AP, and 35% CP (P = .001, χ2; Figure 3). When patients with LBC/Ph+ ALL were subdivided, the frequency was 59% for LBC and 67% for Ph+ ALL.

Frequency of patients with mutations where one or more of their mutations would influence the therapeutic decision. Overall, 43% of our patients with mutations had one or more of the SGI clinically relevant mutations. For patients without T315I, the difference in the frequencies among the disease phases was related to the higher incidence of mutations less sensitive to nilotinib in LBC/Ph+ ALL and AP. The frequency of patients with clinically relevant mutations to nilotinib (Y253H, E255K/V, and F359V/C) or to dasatinib (F317L) is calculated as the percentage of patients without an additional mutation that was less sensitive to both inhibitors.
Frequency of patients with mutations where one or more of their mutations would influence the therapeutic decision. Overall, 43% of our patients with mutations had one or more of the SGI clinically relevant mutations. For patients without T315I, the difference in the frequencies among the disease phases was related to the higher incidence of mutations less sensitive to nilotinib in LBC/Ph+ ALL and AP. The frequency of patients with clinically relevant mutations to nilotinib (Y253H, E255K/V, and F359V/C) or to dasatinib (F317L) is calculated as the percentage of patients without an additional mutation that was less sensitive to both inhibitors.
Our analysis was performed in patients with detectable mutations; and from this, we can estimate the percentage of patients with an SGI clinically relevant mutation among all imatinib-resistant patients. For imatinib-resistant CP patients commencing nilotinib or dasatinib, 48% to 55% had a mutation.36,37 Similarly, for imatinib-resistant patients in AP who commenced nilotinib or dasatinib, the frequency of mutations was 62% to 64%.27,53 Mutations in patients with LBC/Ph+ ALL were detected in 62% to 83%9,28,78 and up to 75% of patients with MBC.9 From these mutation frequencies, the estimated percentage of all imatinib-resistant patients with an SGI clinically relevant mutation is 18% for CP, 31% for AP, up to 29% for MBC, and 39% to 52% for LBC/Ph+ ALL. For imatinib-intolerant CP patients, the frequency of mutations was 8% to 10%,36,37 and these patients will have a considerably lower chance of an SGI clinically relevant mutation. As the clinical data mature, the significance of other mutations, such as Q252H, Y253F, and F359I, for response to nilotinib or dasatinib may become apparent.
Is there a difference in the frequency of multiple mutations among the disease phases?
The prediction of response for patients with more than one mutation may be complicated. Multiple mutations can represent different imatinib-resistant clones or occur on the same BCR-ABL molecule (compound mutations). Direct sequencing techniques will not consistently distinguish these situations. Studies have demonstrated altered transformation potencies when more than one mutation occurred on the same BCR-ABL molecule.50,61 For instance, the dasatinib-resistant T315A mutation displays substantially reduced transformation potency relative to wild-type BCR-ABL in vitro.50,61 However, T315A in the context of other mutations had an increased transformation potency.50,61 Interestingly, a patient who acquired T315A in a BCR-ABL clone with 2 imatinib-resistant mutations (triple compound mutation) responded to a combination of imatinib and dasatinib.50 Furthermore, a compound mutation recovered in a nilotinib resistance screen displayed a cooperative reduction of nilotinib sensitivity compared with both single mutations.43 The data suggest that response and progression for patients treated with kinase inhibitors could be modulated by compound mutations and response is difficult to predict.
We found multiple mutations in 92 of 386 patients (24%). The majority (n = 73) had 2 mutations (80%), 15 had 3 (16%), and 4 had 4 (4%). CP patients had a lower frequency of multiple mutations (17%) compared with other phases: LBC/Ph+ ALL, 27%; MBC, 36%; and AP, 30%. This is consistent with a greater degree of genetic instability in advanced phases.79-82 Interestingly, of the patients with T315I in our cohort, 23 of 53 (43%) also had another mutation. This may be associated with the higher frequency of T315I in advanced phases and a higher frequency of multiple mutations in these phases. Of all patients with an SGI clinically relevant mutation, 35% had multiple mutations (58 of 166 patients). It is currently unknown whether clinical responses to SGIs are modulated when multiple mutations are present: however, the in vitro data suggest that this could be the case for some patients.
Conclusions
A substantial number of patients with imatinib-resistant mutations will achieve a response to SGIs. However, it is apparent from clinical studies that some mutations will indeed influence response to a particular SGI, although expanded clinical studies are required for a definitive set of clinically relevant mutations for each inhibitor. Sensitive mutation assays directed at the detection of these specific mutations may become important. Nevertheless, a search for all mutations is relevant. Among imatinib-resistant patients, the detection of any mutation at the time of commencing SGI therapy may identify those at higher risk of relapse associated with new SGI clinically relevant mutations.36,48 In one study of 95 patients treated with SGI therapy after imatinib failure, 45% of patients already harboring mutations compared with 18% without mutations relapsed with newly acquired mutations.48 In these 45 patients, 98% of the new mutations were those we have classified as SGI clinically relevant. Therefore, patients with mutations at the time of commencing SGI therapy may benefit from regular mutation analysis to allow the early detection of new SGI clinically relevant mutations because these may be associated with loss of response.48
The frequency of mutations that influence the therapeutic decision varies considerably according to the disease phase. In our cohort with mutations at the time of ceasing imatinib and/or at commencement of nilotinib or dasatinib, the incidence of the pan-resistant T315I mutant was up to 3 times lower in CP patients than in advanced disease. Furthermore, imatinib-resistant CP patients had the lowest estimated frequency of SGI clinically relevant mutations (18%). Nevertheless, this indicates that the mutation status in almost one-fifth of imatinib-resistant CP patients should direct therapy away from one or more of the clinically available SGIs. Mutation status will also be relevant for third-line inhibitor therapy. Novel mutations that have emerged with dasatinib resistance, such as V299L, T315A, and F317I,49,50 may be sensitive to nilotinib,50 whereas V299L may be resistant to bosutinib.39
There is still much to learn about the impact of BCR-ABL mutations from continued clinical analysis. Perhaps the strongest evidence for response of mutations to SGIs will come from clinical studies rather than further in vitro testing. It is currently unknown whether all new mutations, other than those identified as SGI clinically relevant, are associated with resistance. Furthermore, it is probable that compound mutations modulate the response to inhibitors in unexpected ways, thereby making prediction of response more complex.
What of first-line nilotinib, dasatinib, or bosutinib therapy, where clinical trials are under way? Will the acquisition of BCR-ABL mutations continue to play a major role in resistance? If so, one could predict that they will be the same mutations that have emerged with resistance in second-line inhibitor therapy. Alternatively, will rare, novel mutations that are resistant to SGIs be detected with longer follow-up? Rare, novel mutations are still detected on occasion in imatinib-treated patients after almost 9 years of analysis.
We now have sufficient evidence to classify a small number of relatively common imatinib-resistant mutations as SGI clinically relevant. Therefore, mutation analysis is required for rational treatment decisions. Not all clinicians will have timely access to mutation analysis for therapeutic decisions, in which case, use of an SGI may be necessary, pending the results of mutation testing. For laboratories performing mutation testing, it is essential that they prioritize mutation analysis for patients with potential or confirmed imatinib failure to aid clinicians in therapeutic decisions.
Authorship
Contribution: S.B wrote the paper; and J.V.M. and T.P.H. reviewed and provided critical revisions to the paper.
Conflict-of-interest disclosure: S.B., J.V.M., and T.P.H. have received research funding and honoraria from Novartis Pharmaceuticals and Bristol-Myers Squibb.
Correspondence: Susan Branford, Department of Molecular Pathology, SA Pathology, Frome Rd, Adelaide SA 5000, Australia; e-mail: susan.branford@health.sa.gov.au.
