

Abstract
Secondary imatinib resistance in chronic myeloid leukemia (CML) is associated in approximately 50% of cases with mutations in the BCR-ABL kinase domain, necessitating switch to one of several new tyrosine kinase inhibitors (TKIs) that act differentially on mutated BCR-ABL. We assess here whether scoring mutation based on in vitro inhibitory concentration of each TKI-mutation pair can predict long-term clinical outcome. Among 169 patients with CML after imatinib failure, mutations were detected before TKI switch in 41 (48%) treated with dasatinib and 45 (52%) treated with nilotinib. Inhibitory concentration values for each TKI-mutation pair were stratified into high (n = 42), intermediate (n = 25), low (T315I, n = 9), or unknown sensitivity (n = 10). Hematologic and cytogenetic response rates were similar for patients with or without mutations. For patients in chronic phase, hematologic and cytogenetic responses correlated with mutation score; tumors with low and intermediate scores had lower response rates than those with highly sensitive mutations, and worse event-free and overall survival. These correlations with overall survival were not seen for advanced phases. Mutation scoring can predict outcome in CML-chronic phase with imatinib failure treated with second-generation TKIs and can help in therapy selection. More complex prognostic models will be required for advanced stages of disease.
Introduction
Therapy with the Bcr-Abl tyrosine kinase inhibitors (TKIs) has revolutionized the management and prognosis in patients with chronic myeloid leukemia (CML).1 Imatinib therapy induced high rates of complete cytogenetic (CCyR) and major molecular responses and improved survival in CML.2-5 After imatinib treatment, more than 90% of patients obtain complete hematologic response, and more than 80% achieve a CCyR. After 6 years of follow-up, the event-free survival (EFS) is 83% and overall survival (OS) nearly 90%, resulting in a major change in the natural history of the disease.6
Despite the significant efficacy of imatinib, some patients may eventually develop resistance,7 with a reported annual resistance rate of less than 1% to 7% in newly diagnosed patients in chronic phase (CP), with the incidence probably decreasing over time.6,8 Mutations in the kinase domain (KD) of BCR-ABL are the most prevalent mechanism of imatinib resistance in patients with CML.9-12
To overcome imatinib resistance, more potent TKIs, such as dasatinib and nilotinib, have been developed, with demonstrable preclinical activity against most imatinib-resistant BCR-ABL KD mutations, with the exception of T315I.13-15 The relative sensitivity of each mutation to different TKIs varies considerably as reflected by inhibitory concentration (IC50) required to inhibit the kinase activity and the proliferation of cells bearing different mutations. The clinical efficacy of the second-generation TKIs has been demonstrated across all phases of CML after imatinib failure in patients with different types of mutations, with high rates of hematologic and cytogenetic responses.16,17
The aims of the study were to investigate whether in vitro sensitivity of KD mutations can be used to predict the response to therapy and, more important, the long-term outcome of patients receiving second-generation TKIs after imatinib failure.
Methods
Between March 2004 and February 2006, 169 of 217 patients (78%) with CML with imatinib failure were evaluated by cDNA sequencing for mutations in the entire KD of BCR-ABL before changing therapy to a second-generation TKI. A kinase domain mutation was identified in 86 (51%) patients. Forty-one patients were subsequently treated with dasatinib, and 45 patients with nilotinib.
The criterion to trigger initial mutation analysis was based on clinical evidence of imatinib failure, as defined in the recent recommendations of the European Leukemia Net.18 Briefly, treatment failure was defined as loss of a cytogenetic or complete hematologic response (CHR), or failure to achieve a CHR (CP only) or any hematologic response (for patients in accelerated phase [AP] or blast phase [BP]) after 3 months of therapy, or persistence of 100% Philadelphia chromosome (Ph)–positive metaphases after 6 months of therapy, or more than or equal to 35% after 12 months.
Patients were registered in protocols approved by the Institutional Review Board of M. D. Anderson Cancer Center and signed an Institutional Review Board–approved informed consent according to institutional guidelines and the Declaration of Helsinki. Response criteria were as previously described.19 A CHR was defined as a white blood cell count of less than 10 × 109/L, a platelet count of less than 450 × 109/L, no immature cells (blasts, promyelocytes, myelocytes) in the peripheral blood, and disappearance of all signs and symptoms related to leukemia (including palpable splenomegaly). A CHR was further categorized by the best cytogenetic response as complete (0% Ph-positive), partial (1%-35% Ph-positive), minor (36%-65% Ph-positive), and minimal (66%-95% Ph-positive). A major cytogenetic remission (MCyR) included complete plus partial cytogenetic responses (ie, Ph-positive ≤ 35%). Cytogenetic response was judged by standard cytogenetic analysis in 20 metaphases done on bone marrow aspiration; fluorescent in situ hybridization, on peripheral blood, was used only when routine cytogenetic analysis was not successful (ie, insufficient metaphases).
Mutation analysis
Total RNA was isolated from peripheral blood or bone marrow aspirate samples by Trizol solubilization (Invitrogen) and cDNA synthesized by reverse transcriptase (Superscript II; Invitrogen). The kinase domain of the BCR-ABL fusion transcript was sequenced using a nested polymerase chain reaction (PCR) strategy. BCR-ABL was first amplified followed by 2 separate PCR reactions that cover codons 221 to 390 and codons 380 to 500 of the ABL KD, respectively. Standard dideoxy chain-termination DNA sequencing was performed using Big Dye chain terminator reagents on an automated 3130 genetic analyzer with analysis by Sequence Analysis, Version 3.3, and SeqScape software, Version 2.5 (Applied Biosystems). All mutations were confirmed by sequencing of forward and reverse strands, with a sensitivity of 10% to 20% mutation-bearing cells in the analyzed population. For analysis of follow-up samples, pyrosequencing was performed after first-round PCR (as mentioned earlier in this paragraph). PCR was performed using one biotin-tagged primers, single-stranded PCR product isolated on strepavidin sepharose beads (GE Healthcare), and sequenced using nucleotide dispensation tips and Pyro Gold reagents on a HSQ96 Pyrosequencer (Biotage). The sensitivity of the pyrosequencing was 1% to 5% mutation-bearing transcripts depending on the initial levels of fusion transcript.
The published IC50 values for each drug for in vitro inhibition (in cell lines) of kinase activity of particular mutated BCR-ABL13,15,20-25 was used to classify mutations into high, intermediate, and low sensitivity to dasatinib (IC50 values ≤ 3 nM, 3-60 nM, and > 60 nM, respectively) and nilotinib (IC50 values ≤ 50 nM, 50-500 nM, and > 500 nM, respectively). Whenever a discrepancy in reported IC50 values was identified between different reports, the worse-case scenario was adopted (ie, the highest IC50 to the corresponding TKI). Compound mutations were classified based on the mutation with the highest IC50.
Statistical analysis
Differences among variables were evaluated by the χ2 test and Mann-Whitney U test for categorical and continuous variables, respectively.26 EFS was measured from the start of a second-generation TKI until loss of the best response (cytogenetic or hematologic) achieved, progression to AP or BP, or death from any cause during treatment. OS was defined from the start of second-generation TKI therapy to the date of death or last follow-up. Survival probabilities were estimated by the Kaplan-Meier method and compared by the log-rank test.27 Univariate and multivariate analyses were performed to identify potential prognostic factors associated with major cytogenetic response and survival. The χ2 test was used to identify prognostic factors, which were then included as variables in a multivariate regression model for response. Factors retaining significance in the multivariate model were interpreted as being independently predictive of major cytogenetic response. Multivariate analysis of survival used the Cox proportional hazard model.27-29
Results
Patient characteristics
Table 1 summarizes the overall patients' characteristics. For patients harboring a KD mutation, median age was 52 years (range, 17-80 years). Fifty-seven patients (66%) had received prior therapy with interferon-α before the start of imatinib; 29 (34%) had received imatinib as their first-line therapy for CML. At the start of imatinib, 68 (79%) patients were in CP, 15 (17%) in AP, and 3 (4%) in BP. Best response to imatinib was CHR only in 43 (50%) patients and major cytogenetic response in 33 (38%; complete in 22 and partial in 11).
Patient characteristics
| Parameter | Mutation group, no. (%) | P | ||
|---|---|---|---|---|
| Mutation (N = 86) | No mutation (N = 83) | |||
| Median age, y (range) | 52 (17-80) | 48 (21-94) | .311 | |
| Prior interferon-α | 57 (66) | 42 (51) | .060 | |
| Stage at imatinib therapy | CP | 68 (79) | 64 (77) | .530 |
| AP | 15 (17) | 13 (16) | ||
| BP | 3 (4) | 6 (7) | ||
| Best response to imatinib | CHR | 43 (50) | 35 (42) | .379 |
| MCyR | 33 (38) | 34 (41) | .881 | |
| CCyR | 22 (26) | 26 (31) | .562 | |
| Median duration of response to imatinib therapy, mo (range) | 25 (2-68) | 29 (3-69) | .20 | |
| Mutation at imatinib failure | Low IC50 | 42 (49) | ||
| Intermediate IC50 | 25 (29) | |||
| High IC50 | 9 (10) | |||
| Unknown IC50 | 10 (12) | NA | NA | |
| Stage at TKI switch | CP | 30 (35) | 29 (35) | .150 |
| AP | 41 (48) | 30 (36) | ||
| BP | 15 (17) | 24 (29) | ||
| Second TKI | Dasatinib | 41 (48) | 40 (48) | .928 |
| Nilotinib | 45 (52) | 43 (52) | ||
| Median follow-up from second TKI switch, mo (range) | 23 (3-38) | 22 (3-36) | .861 | |
| Parameter | Mutation group, no. (%) | P | ||
|---|---|---|---|---|
| Mutation (N = 86) | No mutation (N = 83) | |||
| Median age, y (range) | 52 (17-80) | 48 (21-94) | .311 | |
| Prior interferon-α | 57 (66) | 42 (51) | .060 | |
| Stage at imatinib therapy | CP | 68 (79) | 64 (77) | .530 |
| AP | 15 (17) | 13 (16) | ||
| BP | 3 (4) | 6 (7) | ||
| Best response to imatinib | CHR | 43 (50) | 35 (42) | .379 |
| MCyR | 33 (38) | 34 (41) | .881 | |
| CCyR | 22 (26) | 26 (31) | .562 | |
| Median duration of response to imatinib therapy, mo (range) | 25 (2-68) | 29 (3-69) | .20 | |
| Mutation at imatinib failure | Low IC50 | 42 (49) | ||
| Intermediate IC50 | 25 (29) | |||
| High IC50 | 9 (10) | |||
| Unknown IC50 | 10 (12) | NA | NA | |
| Stage at TKI switch | CP | 30 (35) | 29 (35) | .150 |
| AP | 41 (48) | 30 (36) | ||
| BP | 15 (17) | 24 (29) | ||
| Second TKI | Dasatinib | 41 (48) | 40 (48) | .928 |
| Nilotinib | 45 (52) | 43 (52) | ||
| Median follow-up from second TKI switch, mo (range) | 23 (3-38) | 22 (3-36) | .861 | |
NA indicates not applicable.
Ninety-four mutations were identified in 86 patients with imatinib failure after a median of 25 months (range, 2-68 months) from the start of imatinib therapy and before the start of therapy with a second TKI. Seven patients harbored more than 1 mutation (6 had 2 and 1 had 3). G250E was the most frequent mutation, being present in 14 (16%) patients. As a group, P-loop mutations, defined as mutations in amino acids 248 to 255,18 accounted for 32% of the mutations. T315I was present in 9 patients, representing 10% of all mutations (Table 2).
Mutation distribution
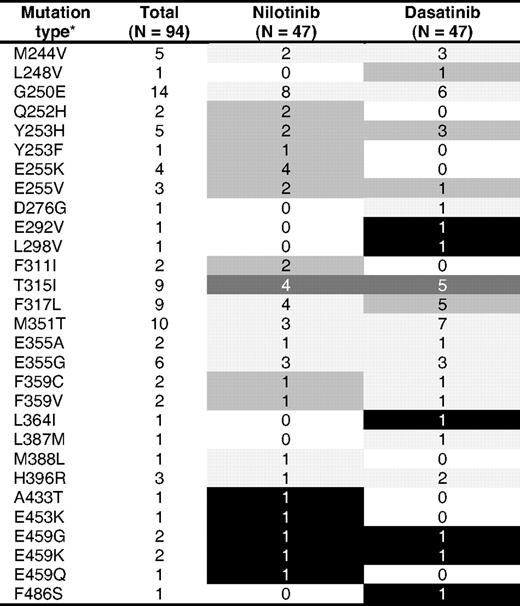
Light gray indicates low IC50; intermediate gray, intermediate IC50; dark gray, high IC50; and black, unknown IC50.
*Seven patients harbored more than 1 mutation (6 harbored 2 and 1 harbored 3).
Forty-two patients (49%) harbored mutations with low IC50 to the inhibitor they received: 21 were treated with dasatinib (10 CP, 7 AP, 4 BP) and 21 with nilotinib (5 CP, 14 AP, 2 BP). Twenty-five patients (29%) harbored intermediate IC50 mutations: 10 were treated with dasatinib (7 CP, 1 AP, 2 BP) and 15 with nilotinib (1 CP, 9 AP, 5 BP). Nine patients (10%) harbored high IC50 mutations (all were T315I mutations): 5 were treated with dasatinib (2 CP, 3 AP) and 4 with nilotinib (2 CP, 1 AP, 1 BP). Ten patients (12%) harbored mutations with no reported in vitro sensitivity: 5 received dasatinib (1 CP, 3 AP, 1 BP) and 5 nilotinib (2 CP, 3 AP). Table 2 summarizes the list of mutations encountered at imatinib failure and before the start of second TKI therapy.
Hematologic and cytogenetic response rates to second-generation TKIs by mutation status
There was no difference in patient characteristics between those with mutations at the time of imatinib failure versus those with no mutations (Table 1). By the time therapy with the second TKI was initiated for patients with mutations, 38 patients treated with imatinib while in CP had transformed to AP or BP, whereas 30 patients still remained in CP. Forty-one patients (20 CP, 14 AP, and 7 BP) received dasatinib and 45 (10 CP, 27 AP, and 8 BP) received nilotinib after developing failure to imatinib therapy. The median follow-up from second-generation TKI switch was 23 months (range, 3-38 months).
Hematologic and cytogenetic response rates were similar for patients without or with KD mutations (Table 3). There was a trend for lower hematologic response rates for patients in CP harboring mutations, with 77% of patients in CP with baseline mutation after imatinib failure achieving a complete hematologic response with second TKIs versus 96% for patients without KD mutations (P = .055); 58% of patients with KD mutations achieved a MCyR versus 68% for those without KD mutations (P = .43); and 52% achieved a CCyR versus 54% for those without KD mutations (P = .88). Response rates were higher in patients with low IC50 mutations compared with patients with intermediate IC50 mutations. The rates of MCyR and CCyR were, respectively, 87% and 73% versus 25% (P = .003) and 25% (P = .026).
Responses by mutation types and disease phase
| Phase/mutation status before second TKI | N | Response, % | ||
|---|---|---|---|---|
| CHR | MCyR | CCyR | ||
| CP | ||||
| None | 29 | 96 | 68 | 54 |
| Mutated | 30 | 77 | 58 | 52 |
| IC50 low* | 15 | — | 87 | 73 |
| IC50 intermediate* | 8 | — | 25 | 25 |
| P† | .003 | .026 | ||
| AP | ||||
| None | 30 | 70 | 30 | 20 |
| Mutated | 41 | 68 | 34 | 31 |
| IC50 low | 21 | — | 38 | 38 |
| IC50 intermediate | 10 | — | 10 | 10 |
| P† | .01 | .01 | ||
| BP | ||||
| None | 24 | 37 | 25 | 21 |
| Mutated | 15 | 27 | 20 | 13 |
| IC50 low | 6 | — | 33 | 17 |
| IC50 intermediate | 7 | — | 14 | 14 |
| P† | .42 | .9 | ||
| Phase/mutation status before second TKI | N | Response, % | ||
|---|---|---|---|---|
| CHR | MCyR | CCyR | ||
| CP | ||||
| None | 29 | 96 | 68 | 54 |
| Mutated | 30 | 77 | 58 | 52 |
| IC50 low* | 15 | — | 87 | 73 |
| IC50 intermediate* | 8 | — | 25 | 25 |
| P† | .003 | .026 | ||
| AP | ||||
| None | 30 | 70 | 30 | 20 |
| Mutated | 41 | 68 | 34 | 31 |
| IC50 low | 21 | — | 38 | 38 |
| IC50 intermediate | 10 | — | 10 | 10 |
| P† | .01 | .01 | ||
| BP | ||||
| None | 24 | 37 | 25 | 21 |
| Mutated | 15 | 27 | 20 | 13 |
| IC50 low | 6 | — | 33 | 17 |
| IC50 intermediate | 7 | — | 14 | 14 |
| P† | .42 | .9 | ||
— indicates not reported.
In vitro IC50 for a given TKI for a given mutation.
Comparing low versus intermediate mutation classes.
Sixty-eight percent of patients with baseline mutations in AP achieved a CHR versus 70% for patients without KD mutations (P = .87); 34% of patients with KD mutations achieved a MCyR (complete in 31%) versus 30% for those without KD mutations (complete in 20%; P = .71 and .27, respectively). Response rates were higher in patients with low IC50 mutations compared with patients with intermediate IC50 mutations. The rates of MCyR and CCyR (all MCyR were complete) were, respectively, 38% and 10% (P = .01). There was no difference in response rates between patients with or without KD mutations in BP. The rates of CHR, MCyR, and CCyR were, respectively, 27%, 20%, and 13% versus 37% (P = .68), 25% (P = .72), and 21% (P = .55). There was only a trend for lower response rates for patients with mutations with higher IC50. Overall, there was no difference in the time to achievement of a MCyR among patients with or without mutations regardless of the IC50 of the mutations (data not shown).
All 10 patients (3 CP, 6 AP, 1 BP) harboring mutation with no reported sensitivity responded. All 3 patients in CP achieved CCyR that was lost in one (A433T treated with nilotinib) after 6 months and was sustained in 2 for 25+ (L298V treated with dasatinib) and 26+ (E459K treated with nilotinib) months. Of the 6 patients in AP, 1 patient (F486S treated with dasatinib) achieved CHR only, sustained for 24+ months, and 5 (3 with E453K, E459Q, and E459G mutation each, were treated with nilotinib and 2 with E459K and L364I mutation each, were treated with dasatinib) achieved MCyR (complete in 4) sustained for a median of 23+ months (range, 12-33 months). Only 1 patient (with L364I mutation treated with dasatinib) lost CCyR after acquiring a F317L mutation 32 months into therapy. The patient in BP (E292V treated with dasatinib) achieved a transient return to second CP of 3 months duration, then progressed and died.
Of the 7 patients with compound mutations, 1 patient did not respond to second-generation TKI, 3 achieved CHR only, lost after a median of 8 months (range, 5-12 months), and 3 patients a MCyR (complete in 2) lost after a median of 11 months (range, 6-17 months). Of the 83 patients with no baseline mutation at the second TKI switch, 49 had sequential cDNA sequencing. Seventeen patients (35%) acquired new mutations that were associated with clinical TKI resistance in 82% of the cases. Of the 86 patients with KD mutations at the TKI switch, 59 had sequential cDNA sequencing. Seven patients (12%) acquired new mutations that were associated with TKI resistance in 86%.
Outcome and survival by mutation status
After a median follow-up of 23 months from the start of therapy with second-generation TKIs, 51 events were reported among 83 patients (61%) without baseline mutations (9 in CP, 21 in AP and BP, each) and 59 among 86 patients (69%) with baseline mutations (14 in CP, 31 in AP, 14 in BP). Of the 83 patients without baseline mutations, 48 (58%) were alive compared with 52 of 86 (60%) with any KD mutation.
Table 4 summarizes outcome by phase and type of mutations. In CP, the 2-year EFS rates for patients with no mutation versus those with low, intermediate, high, or not reported IC50 mutations, were 63% compared with 78%, 22%, 0%, and 67%, respectively (Table 4). Although there was no difference between patients with no mutations or low IC50 mutations (P = .58), patients with intermediate IC50 mutations had a significant worse outcome (P = .001; Figure 1A). This difference impacted survival of patients in CP. The 2-year overall survival rates were 96%, 100%, 70%, 75%, and 100%, respectively. Similarly, although there was no difference in overall survival between patients with no or low IC50 mutations (P = .74), those with intermediate IC50 mutations had worse outcome (P = .03; Figure 1B).
Outcome by mutation types and disease phase
| Phase/mutation status before second TKI | N | 2-year, % | |
|---|---|---|---|
| EFS | OS | ||
| CP | |||
| None | 29 | 63 | 96 |
| Mutated low IC50 | 15 | 78 | 100 |
| Mutated intermediate IC50 | 8 | 22 | 70 |
| Mutated high IC50 | 4 | 0 | 75 |
| Mutated unknown IC50 | 3 | 67 | 100 |
| P* | <.001 | .03 | |
| AP | |||
| None | 30 | 27 | 49 |
| Mutated low IC50 | 21 | 9 | 60 |
| Mutated intermediate IC50 | 10 | 11 | 42 |
| Mutated high IC50 | 4 | 0 | 50 |
| Mutated unknown IC50 | 6 | 84 | 80 |
| P* | .3 | .37 | |
| BP | |||
| None | 24 | 23 | 29 |
| Mutated low IC50 | 6 | 20 | 14 |
| Mutated intermediate IC50 | 7 | 0 | 14 |
| Mutated high IC50 | 1 | 0 | 0 |
| Mutated unknown IC50 | 1 | 0 | 0 |
| P* | .7 | .17 | |
| Phase/mutation status before second TKI | N | 2-year, % | |
|---|---|---|---|
| EFS | OS | ||
| CP | |||
| None | 29 | 63 | 96 |
| Mutated low IC50 | 15 | 78 | 100 |
| Mutated intermediate IC50 | 8 | 22 | 70 |
| Mutated high IC50 | 4 | 0 | 75 |
| Mutated unknown IC50 | 3 | 67 | 100 |
| P* | <.001 | .03 | |
| AP | |||
| None | 30 | 27 | 49 |
| Mutated low IC50 | 21 | 9 | 60 |
| Mutated intermediate IC50 | 10 | 11 | 42 |
| Mutated high IC50 | 4 | 0 | 50 |
| Mutated unknown IC50 | 6 | 84 | 80 |
| P* | .3 | .37 | |
| BP | |||
| None | 24 | 23 | 29 |
| Mutated low IC50 | 6 | 20 | 14 |
| Mutated intermediate IC50 | 7 | 0 | 14 |
| Mutated high IC50 | 1 | 0 | 0 |
| Mutated unknown IC50 | 1 | 0 | 0 |
| P* | .7 | .17 | |
Comparing low versus intermediate mutation classes.

Event-free and overall survival for all patients in chronic phase. (A) Event-free survival by IC50 in chronic phase. (B) Overall survival by IC50 in chronic phase.
Event-free and overall survival for all patients in chronic phase. (A) Event-free survival by IC50 in chronic phase. (B) Overall survival by IC50 in chronic phase.
Among those in AP before TKI switch, the 2-year EFS rates for patients without mutations versus those with low, intermediate, high, or not reported IC50 mutations were 27%, 9%, 11%, 0%, and 84%, respectively (Table 4). The 2-year OS rates were 49%, 60%, 42%, 50%, and 80%, respectively. Thus in AP, the presence of a mutation or its predicted sensitivity had minimal impact on the EFS and OS (Figure 2). Among those patients who had already progressed to BP before TKI switch, the median EFS of patients without mutations, low, intermediate, high, and not reported IC50 mutations was 3, 7, 2, 2, and 1 months, respectively. The 2-year EFS rates were 23%, 20%, 0%, 0%, and 0%, respectively. The median OS was, respectively, 6, 11, 5, 4, and 4 months. The 2-year OS rates were 29%, 14%, 14%, 0%, and 0%, respectively (Table 4). Similar to AP disease, neither the presence of mutations nor the IC50 of the mutations impacted significantly the EFS and OS.

Event-free and overall survival for all patients in accelerated phase. (A) Event-free survival by IC50 in accelerated phase. (B) Overall survival by IC50 in accelerated phase.
Event-free and overall survival for all patients in accelerated phase. (A) Event-free survival by IC50 in accelerated phase. (B) Overall survival by IC50 in accelerated phase.
Prognostic factors for major cytogenetic response and survival
We then analyzed the characteristics that might be associated with the achievement of MCyR after therapy with second-generation TKIs, long-term EFS, and OS. Factors analyzed were: age, sex, time from diagnosis, prior interferon therapy, previous response to imatinib therapy, stage at the start of second-generation TKI therapy, clonal evolution, and type of mutations. By multivariate analysis, the stage of the disease (CP > AP > BP) at the start of second-generation TKI, the absence of intermediate or high IC50 mutations (compared with no or low IC50 mutations), and a previous MCyR to imatinib therapy were independently associated with the achievement of MCyR to second-generation TKIs and longer EFS and OS (Table 5). Clonal evolution had no independent impact on the achievement of a MCyR after therapy with second-generation TKI, nor on EFS and OS.
Multivariate analysis
| Positive features | MCyR | EFS | OS | |||
|---|---|---|---|---|---|---|
| Odds ratio | P | Hazard ratio | P | Hazard ratio | P | |
| Chronic phase | 0.29 | < .001 | 2.2 | < .001 | 4.2 | < .001 |
| No or low IC50 mutation | 0.18 | .003 | 2.1 | .002 | 1.8 | .06 |
| Previous MCyR to imatinib | 7.5 | < .001 | 0.56 | .006 | 0.56 | .04 |
| Positive features | MCyR | EFS | OS | |||
|---|---|---|---|---|---|---|
| Odds ratio | P | Hazard ratio | P | Hazard ratio | P | |
| Chronic phase | 0.29 | < .001 | 2.2 | < .001 | 4.2 | < .001 |
| No or low IC50 mutation | 0.18 | .003 | 2.1 | .002 | 1.8 | .06 |
| Previous MCyR to imatinib | 7.5 | < .001 | 0.56 | .006 | 0.56 | .04 |
Discussion
The availability of second-generation TKIs has provided new therapeutic options for patients with imatinib resistance. These agents have in vitro activity against all imatinib-resistant KD mutants, except T315I.13-15 The relative sensitivity of each mutation to different TKIs varies considerably as reflected by the IC50 determined by in vitro kinase inhibition assays. In our study, we assessed whether scoring mutation based on in vitro IC50 of each TKI-mutation pair can predict long-term clinical outcome.
The clinical efficacy of second-generation TKIs has been demonstrated across all phases of CML after imatinib failure, with high rates of hematologic and cytogenetic responses.16,17 Our study confirmed and extended these findings in that: (1) second-generation TKI treatment resulted in hematologic and cytogenetic responses in a significant proportion of imatinib-resistant patients with BCR-ABL mutations; (2) patients with no baseline mutations and those with mutations of low IC50 had similar response and progression rates, whereas those with baseline mutations of intermediate and high IC50 had lower response rates and a higher risk of progression; and (3) the correlation with IC50 was less evident in advanced phases.
The finding that second-generation TKIs showed equivalent efficacy in patients with and without baseline mutations across all CML phases is strong evidence of the ability of the second-generation TKIs to effectively suppress BCR-ABL-directed cell proliferation with a wide variety of mutants.15,20 This is most clearly demonstrated by the similar cytogenetic response rates in these 2 groups: approximately 60% for patients in CP, 30% for patients in AP, and 25% for those in BP. This is in line with the results of the phase 2 pivotal trials in all phases.30-35 The outcome was particularly similar for patients in CP with no or low IC50 mutations, with equivalent rates of hematologic and cytogenetic responses and similar 2-year EFS and OS. Similar findings were recently reported in abstract form by other investigators.31,36-38
In contrast, patients with intermediate IC50 had lower response rates and worse outcome with significantly lower 2-year EFS and OS. In our study, patients in CP with intermediate IC50 mutations had shorter duration of response and worse survival compared with those with no or low IC50 mutations. In keeping with this observation, many of the intermediate IC50 mutations have been implicated in primary or secondary clinical resistance to the second-generation TKIs, in smaller series.39-41
We and other investigators have reported that dasatinib is effective in patients who have failed nilotinib, and vice versa.42,43 This is particularly true if the patient has certain mutations.44 For example, nilotinib is more active than dasatinib in patients with F317L mutations, whereas dasatinib is more active in patients with mutations, such as some P-loop mutations and F359V. These differences lend support to the selection of a particular TKI over the other based on the in vitro sensitivity of the specific mutation to each TKI.
Interestingly, among patients with advanced phases, the in vitro sensitivity of the mutations had minimal or no impact on EFS and OS. This can be explained in part by the fact that resistance in advanced phases is multifactorial, frequently accompanied by additional chromosomal abnormalities, and possibly the activation of other oncogenic pathways.9,45 In addition, the cutoffs that define sensitivity in vitro are somewhat arbitrary. Therefore, treatment strategies that pair BCR-ABL kinase inhibition with other targeted therapy are needed.
Finally, in line with what we had previously reported,39 the acquisition of a new mutation after therapy with second-generation TKI was associated with resistance in the majority of the cases. The development of new mutations was more commonly observed among patients not harboring any mutation at the start of therapy with second-generation TKI. This is in contrast with recent reports in large series with dasatinib38 and nilotinib.36 However, those series, besides dealing with individual agents, include only patients in chronic phase, while our report includes patients in all stages of the disease. New mutations in patients with no pre-existing mutations were more frequently seen among patients treated in advanced stages, with 15 of 17 (88%) of these mutations occurring in patients treated in accelerated or blast phase.
In summary, second-generation TKIs are capable of inducing hematologic and cytogenetic responses in a significant proportion of imatinib-resistant patients across all phases. Outcome depends on the type of mutation, with those mutations with predicted intermediate levels of sensitivity having decreased probability of response and shorter EFS, particularly in CP. The correlation with IC50 is not evident in advanced phases, suggesting more complex mechanisms of resistance. Nevertheless, selection of the subsequent TKI agent should be based not only on the mutation type, but also on other patient and disease features, such as disease stage and comorbidities.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: E.J. analyzed data, wrote the paper, and approved the manuscript; D.J. performed DNA sequencing, wrote the paper, and approved the manuscript; H.M.K. wrote the paper and approved the manuscript; S.O., C.K., J.A.B., G.B., and W.G.W. treated patients and approved the manuscript; C.T. analyzed data; and J.C. analyzed data, wrote the paper, and approved the manuscript.
Conflict-of-interest disclosure: E.J. is a member of the speakers' bureau of Bristol Myers Squibb and Novartis. H.M.K. and J.C. have received research grants from Novartis, Bristol-Myers Squibb, and Wyeth. The remaining authors declare no competing financial interests.
Correspondence: Jorge Cortes, Department of Leukemia, University of Texas, M. D. Anderson Cancer Center, 1515 Holcombe Blvd, Box 428, Houston, TX 77030; e-mail: jcortes@mdanderson.org.
