

Abstract
A unique pentasaccharide fragment of heparin can enhance the reactivity of antithrombin with coagulation proteases factors IXa and Xa by 300- to 600-fold through a conformational activation of the serpin, without having a significant effect on the reactivity of antithrombin with thrombin. In this study, it was hypothesized that differences in the structure of the autolysis loop of coagulation proteases (residues 143-154 in chymotrypsin numbering) may be responsible for their differential reactivity with the native and heparin-activated antithrombin. To test this hypothesis, the autolysis loops of both thrombin and the anticoagulant serine protease-activated protein C were replaced with the corresponding loop of factor Xa. Inhibition studies revealed that in contrast to the approximately 1.5-fold difference in the reactivity of thrombin with antithrombin in the absence and presence of pentasaccharide, the difference in reactivity was increased to approximately 37-fold for the mutant thrombin. In the case of the activated protein C mutant, similar to factor Xa, pentasaccharide accelerated the reaction 375-fold. These results suggest that structural differences in the autolysis loop of coagulation proteases play a key role in their differential reactivity with the native and heparin-activated conformations of antithrombin. (Blood. 2004;104:1753-1759)
Introduction
Antithrombin (AT) is a serine protease inhibitor of the serpin superfamily that regulates the proteolytic activities of the procoagulant proteases of both the intrinsic and extrinsic blood coagulation cascade.1-3 The most important physiologic targets of AT, however, appear to be thrombin, factor IXa (fIXa), and factor Xa (fXa). AT is a slow inhibitor of coagulation proteases unless it is bound to heparin-like glycosaminoglycans that line the microvasculature.2,4 This is the basis for the extensive use of heparin in the treatment of venous thrombosis. Full-length high-affinity heparins are known to accelerate the AT inhibition of coagulation proteases by more than 3 to 4 orders of magnitude.3,5 Such a dramatic heparin-mediated increase in the rate of AT reaction with coagulation proteases is believed to arise from (1) the ability of heparin to change the conformation of the serpin to facilitate its optimal recognition by the enzymes (activation mechanism), and (2) the ability of full-length heparins to bridge the serpin and enzyme in 1 complex, promoting the initial interaction between the 2 proteins (template mechanism). In the case of thrombin, the cofactor effect of a full-length heparin is mediated primarily through a template mechanism with less than a 2-fold contribution from the activation mechanism.3,6 In the case of fIXa and fXa, however, the cofactor effect of a full-length heparin is mediated by approximately 300- to 600-fold enhancement through activation and approximately 200- to 300-fold enhancement through the template mechanism in the presence of physiologic concentrations of Ca2+.7-11 These distinct inhibition mechanisms have been supported by the observation that a unique pentasaccharide fragment of heparin that can bind and activate AT, but is not long enough to bridge the inhibitor and enzyme, specifically promotes the AT inhibition of fXa 200- to 300-fold but has minimal effect on the inhibition of thrombin.6,12
The mechanism by which fIXa and fXa recognize the activated conformation of AT is not known. Recent structural and mutagenesis data have indicated that the binding of heparin to a basic site of AT results in a conformational change in the structure of the serpin that is associated with the exposure of a new interactive site for both fIXa and fXa.2,11,13,14 In support of this hypothesis, we recently demonstrated that the conserved autolysis loop residue Arg150 in both fIXa and fXa may interact with AT specifically in the presence of pentasaccharide.15,16 This was evidenced by the observation that the substitution of Arg150 with Ala resulted in an order of magnitude impairment in the reactivity of the mutants with AT in the presence of pentasaccharide.15,16 Comparison of the sequence of the autolysis loops of coagulation proteases from residues 143-154 (chymotrypsinogen numbering17 ) suggests that this loop has 11 residues in both fIXa and fXa but it is longer by 5 to 6 residues in both thrombin and the nontarget protease-activated protein C (APC).17-20 To test the hypothesis that differences in the structure of the autolysis loops are primarily responsible for the differential reactivity of coagulation proteases with the native and activated conformations of AT, we prepared mutants of both APC and thrombin in which all residues of the autolysis loop (residues 143-154) were replaced with the corresponding residues of fXa. Wild-type APC was unreactive with AT; however, the APC mutant was inhibited by AT with second-order association rate constants of 3.2 M-1s-1 and 1.2 × 103 M-1s-1 in the absence and presence of pentasaccharide, respectively, suggesting a 375-fold enhanced reactivity for the mutant with the activated conformation of the serpin. Similarly, in contrast to approximately 1.5-fold enhanced reactivity of wild-type thrombin with AT in the presence of pentasaccharide, the reactivity of the thrombin mutant with AT was improved approximately 37-fold in the presence of the cofactor. We conclude that structural differences in the autolysis loop of coagulation proteases may primarily be responsible for their differential reactivity with the native and activated conformations of AT.
Materials and methods
Construction, expression, and purification of recombinant proteins
Construction and expression of wild-type protein C in HEK293 cells and prethrombin-2 (prothrombin lacking γ-carboxyglutamic acid and both kringles 1 and 2 domains) in baby hamster kidney cells was described previously.21,22 The protein C and prethrombin-2 mutants containing residues 143-154 (chymotrypsinogen numbering17 ) of factor X (PC-fX143-154 and Pre-2-fX143-154) were constructed by polymerase chain reaction (PCR) mutagenesis methods in the same vector systems as described.21,22 Following confirmation of the accuracy of mutagenesis, the mutant constructs were expressed in HEK293 and baby hamster kidney cells as described.21,22 Both wild-type and mutant zymogens were purified to homogeneity by immunoaffinity chromatography using the Ca2+-dependent monoclonal antibody, called HPC4, coupled to Affi-Gel 10 (Bio-Rad, Hercules, CA) as described.21,22
Human plasma proteins, APC, AT, and factors Xa and Va were purchased from Hematologic Technologies Inc (Essex Junction, VT). The active AT-binding pentasaccharide fragment of heparin (fondaparinux sodium) was purchased from Quintiles Clinical Supplies (Mt Laurel, NJ). A full-length high-affinity heparin with an average molecular mass of approximately 21 000 (∼70 saccharides) was a generous gift from Dr Steven Olson (University of Illinois at Chicago). Concentrations of heparins were based on the AT-binding sites and were determined by stoichiometric titration of AT with polysaccharides, with monitoring of the interaction by changes in protein fluorescence as described.23 The thrombomodulin fragment containing epidermal growth factor-like domains 456 (TM456)24 and protein C inhibitor21 were expressed in mammalian cells by the cited methods. Phospholipid vesicles containing 80% phosphatidylcholine and 20% phosphatidylserine (PC/PS) were prepared as described.25 The chromogenic substrate, Spectrozyme PCa (SpPCa), was purchased from American Diagnostica (Greenwich, CT); S2238 and S2266 were purchased from Kabi Pharmacia/Chromogenix (Franklin, OH). Polybrene, paminobenzamidine, and Oxyuranus scutellatus venom (taipan venom) were purchased from Sigma (St Louis, MO).
Zymogen activation
Following purification on the immunoaffinity column, 2 mg of the mutant protein C was incubated with thrombin (25 μg) in 0.1 M NaCl, 0.02 M Tris-HCl, pH 7.4 (Tris-buffered saline; TBS) containing 5 mM EDTA (ethylenediaminetetraacetic acid) for 2 hours at 37°C. The APC mutant was separated from thrombin by a fast-protein liquid chromatography (FPLC) Mono Q column developed with a 40-mL linear gradient from 0.1 to 1.0 M NaCl, 0.02 M Tris-HCl, pH 7.4 as described.21 The concentrations of wild-type and mutant APC were determined from the absorbance at 280 nm assuming a molecular mass of approximately 56 kDa and extinction coefficient (
The concentration dependence of wild-type and mutant protein C activation by the thrombin-TM456 complex was also compared. In this case, human thrombin (1 nM) in complex with TM456 (100 nM) was incubated with increasing concentrations of protein C (0.06-8.0 μM) in TBS containing 1 mg/mL bovine serum albumin, 0.1% polyethylene glycol 8000, and 2.5 mM CaCl2 (TBS/Ca2+). Following 8 minutes incubation at room temperature, thrombin in the activation reaction was inhibited by the addition of hirudin (20 nM), and the rate of APC generation was calculated by an amidolytic activity assay using SpPCa (200 μM final). The concentrations of APC in the reaction mixtures were determined by reference to standard curves, which were prepared by the total activation of protein C with excess thrombin at the time of the experiments as described.24
Both wild-type and mutant prethrombin-2 (2 mg each) were activated to thrombin by taipan venom (25 μg) in TBS/Ca2+ for 1 hour at 37°C and purified by Mono S ion-exchange chromatography as described.27 The concentrations of wild-type and mutant thrombins were determined from the absorbance at 280 nm assuming a molecular mass of approximately 36.6 kDa and an
The concentration dependence of wild-type and mutant prethrombin-2 activation by fXa was also compared in the presence of factor Va. In this case, fXa (0.1 nM) was incubated with a saturating concentration of factor Va (20 nM) and increasing concentrations of prethrombin-2 (0.1-15 μM) for 2 to 5 minutes at room temperature in TBS/Ca2+. The activation reactions were terminated by the addition of EDTA to a final concentration of 20 mM, and the rate of thrombin generation was measured by an amidolytic activity assay using S2238 as described.22 It was ensured that less than 15% substrate was activated at all concentrations of the zymogen.
Kinetic methods
The rate of inactivation of wild-type and mutant proteases by AT in both the absence and presence of heparin cofactors were measured under pseudo-first-order rate conditions by a discontinuous assay method as described.7 In the absence of a cofactor, both wild-type and mutant proteases (1 nM for thrombin and 2 nM for APC) were incubated with plasma AT (0.125-10 μM) in TBS/Ca2+ at room temperature. In the presence of pentasaccharide (H5), the reaction conditions were the same except that wild-type and mutant proteases were incubated with AT (0.025-10 μM) in the presence of saturating concentrations of H5 (1-20 μM) in TBS/Ca2+. In the presence of a full-length high-affinity heparin (∼70 saccharides), the reaction conditions for APC derivatives were the same except that each protease was incubated with 0.25 to 5 μM AT and catalytic levels of heparin (0.01-2.5 μM). In the case of thrombin, both wild-type and mutant proteases (0.5 nM each) were incubated with 200 nM AT and catalytic levels of heparin (0.5-2.0 nM). All reactions were carried out in 50-μL volumes in 96-well polystyrene plates at room temperature. After a period of time (10 seconds to 300 minutes, depending on the rate of reactions), 50 μL of the chromogenic substrate (400 μM SpPCa) in TBS containing 1 mg/mL polybrene was added to each well, and the remaining enzyme activity was measured with a Vmax Kinetics Microplate Reader (Molecular Devices, Menlo Park, CA). The observed pseudo-first-order rate constants (kobs) were determined by computer fitting of the time-dependent change of the protease activity to a first-order rate equation, and the second-order association rate constants for both uncatalyzed and catalyzed reactions were obtained from the slopes of linear plots of kobs versus the concentration of AT or the AT-heparin complex as described.23 In inhibition reactions where the kobs values exhibited a saturable dependence on the concentration of the AT-heparin complex, data were analyzed according a hyperbolic equation as described.23
Cleavage of chromogenic substrates
The steady-state kinetics of hydrolysis of SpPCa (15-2000 μM for APC and 0.75-100 μM for thrombin) by both wild-type and mutant APC (5 nM) or thrombin (0.5 nM) was measured in TBS/Ca2+ at 405 nm at room temperature in a Vmax kinetic microplate reader. The Km and kcat values for the substrate hydrolysis were calculated from the Michaelis-Menten equation as described.21,28
Inhibition by p-aminobenzamidine (PAB)
The affinity of PAB interaction with the active-site pocket of wild-type and mutant proteases was also evaluated. In all cases, APC and thrombin derivatives (5 nM) were incubated with increasing concentrations of PAB (0-320 μM) in the presence of different fixed concentrations of S2266 (62.5-1000 μM) in TBS/Ca2+. The activities of enzymes were measured from the cleavage rate of the chromogenic substrate as described, and the Ki values were determined by global fitting of data to a competitive binding equation as described.29
Analysis of the protease-AT reaction products by gel electrophoresis
The complex formation of proteases with AT in both the absence and presence of heparin cofactors was monitored by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The inhibition reactions were carried out in 20-μL volumes with equal concentrations of each protease and AT (2 μM) in the absence and presence of either pentasaccharide or high-affinity heparin (4 μM each). Following incubation at 37°C for 10 to 30 minutes, 5 μL of 5 × nonreducing sample buffer was added, boiled for 5 minutes, and each reaction was analyzed by SDS-PAGE on 10% polyacrylamide gel stained with GelCode blue stain reagent (Pierce, Rockford, IL).
Results
Expression and purification of protein C
Both wild-type and mutant protein C were expressed in HEK293 cells and purified to homogeneity on the HPC4 immunoaffinity column as described.21 Previous SDS-PAGE analysis of both plasma and recombinant human protein C under nonreducing conditions has indicated that they both migrate as 2 or 3 subforms with slightly different relative molecular masses (∼50-60 kDa) representing differentially glycosylated variants of the protein.30 The protein C mutant, however, migrated slightly faster than wild-type protein due to the loss of 1 of the 4 potential glycosylation sites of the protein30 (data for the zymogen form is not shown but see Figure 1 lane 3 for the activated form). Relative to the autolysis loop of fXa (143Arg-Thr-His-Glu-Lys-Gly-Arg-Gln-Ser-Thr-Arg154), the corresponding sequence of APC (143Tyr-His-Ser-Ser-Arg-Glu-Lys-Glu-Ala-Lys-Arg-Asn-Arg-Thr-Phe-Val154) is longer by 5 residues and also harbors a known glycosylation site (Asn150, chymotrypsinogen numbering17 ; Asn313, mature protein C numbering31 ). Previous mutagenesis studies have indicated that Asn150 is always glycosylated in protein C,30 thus its deletion resulted in a mutant that migrated slightly faster than the wild-type protein.

Nonreducing SDS-PAGE analysis of the stable AT-protease complex formation with wild-type and mutant APC. An equimolar concentration (2 μM) of either wild-type (lanes 4-6) or mutant APC (lanes 7-9) was incubated with AT for 30 minutes in the absence (lanes 4 and 7) or presence of 4 μM pentasaccharide (lanes 5 and 8) or the approximately 70-saccharide heparin (lanes 6 and 9) in 20-μL reactions in TBS/Ca2+. Five microliters of 5 × nonreducing sample buffer was added to each reaction and the samples were boiled for 5 minutes and applied on 10% SDS gel. The stable mutant protease-AT complexes migrated as high-molecular-weight bands on lanes 8 and 9. The free forms of AT, wild-type, and mutant APC are shown in lanes 1-3, respectively. Lane 10 is molecular mass standard in kDa.
Nonreducing SDS-PAGE analysis of the stable AT-protease complex formation with wild-type and mutant APC. An equimolar concentration (2 μM) of either wild-type (lanes 4-6) or mutant APC (lanes 7-9) was incubated with AT for 30 minutes in the absence (lanes 4 and 7) or presence of 4 μM pentasaccharide (lanes 5 and 8) or the approximately 70-saccharide heparin (lanes 6 and 9) in 20-μL reactions in TBS/Ca2+. Five microliters of 5 × nonreducing sample buffer was added to each reaction and the samples were boiled for 5 minutes and applied on 10% SDS gel. The stable mutant protease-AT complexes migrated as high-molecular-weight bands on lanes 8 and 9. The free forms of AT, wild-type, and mutant APC are shown in lanes 1-3, respectively. Lane 10 is molecular mass standard in kDa.
Protein C activation by thrombin
The protein C mutant was activated to APC by thrombin in the presence of EDTA and purified to homogeneity as described.21 The concentrations of both wild-type and mutant APC were determined by stoichiometric titration of enzymes with known concentrations of protein C inhibitor as described.21 These values for both enzymes correlated well (within 90%-100%) with the concentrations of proteins determined based on the absorbance at 280 nm assuming a molecular weight of approximately 56 kDa and an extinction coefficient (

Concentration dependence of wild-type and mutant protein C and prethrombin-2 activation by the activation complexes. (A) The activation of both wild-type (○) and mutant protein C (•) by thrombin (1 nM) in complex with saturating concentrations of TM456 (100 nM) was monitored in TBS/Ca2+. Following 8 minutes of activation at room temperature, thrombin activity was neutralized by hirudin and the rate of APC generation was determined by an amidolytic activity assay described in “Materials and methods.” (B) The activation of both wild-type (○) and mutant prethrombin-2 (•) by fXa (0.1 nM) in complex with saturating concentrations of factor Va (20 nM) on PC/PS vesicles was monitored in TBS/Ca2+. Following 2 to 5 minutes of activation at room temperature, EDTA was added to a final concentration of 20 mM and the rate of thrombin generation was determined by an amidolytic activity assay described in “Materials and methods.” Solid lines in both panels are nonlinear regression analyses of kinetic data according to the Michaelis-Menten equation.
Concentration dependence of wild-type and mutant protein C and prethrombin-2 activation by the activation complexes. (A) The activation of both wild-type (○) and mutant protein C (•) by thrombin (1 nM) in complex with saturating concentrations of TM456 (100 nM) was monitored in TBS/Ca2+. Following 8 minutes of activation at room temperature, thrombin activity was neutralized by hirudin and the rate of APC generation was determined by an amidolytic activity assay described in “Materials and methods.” (B) The activation of both wild-type (○) and mutant prethrombin-2 (•) by fXa (0.1 nM) in complex with saturating concentrations of factor Va (20 nM) on PC/PS vesicles was monitored in TBS/Ca2+. Following 2 to 5 minutes of activation at room temperature, EDTA was added to a final concentration of 20 mM and the rate of thrombin generation was determined by an amidolytic activity assay described in “Materials and methods.” Solid lines in both panels are nonlinear regression analyses of kinetic data according to the Michaelis-Menten equation.
Expression, purification, and activation of prethrombin-2
Both wild-type and mutant prethrombin-2 were expressed in baby hamster kidney cells and purified to homogeneity as described.22 The prethrombin-2 zymogens were converted to thrombin by taipan venom and purified on a Mono S ion-exchange column as described.28 SDS-PAGE analysis under nonreducing conditions suggested that both wild-type and mutant thrombins were purified to homogeneity and that they both migrated at expected relative molecular masses of approximately 37 kDa (data not shown). Relative to fXa, the autolysis loop of thrombin (143Asn-Leu-Lys-Glu-Thr-Trp-Thr-Ala-Asn-Val-Gly-Lys-Gly-Gln-Pro-Ser-Val154) is longer by 6 residues. To determine whether the autolysis loop substitution influences the zymogenic property of the mutant protein in the prothrombinase complex, the concentration dependence of the activation of wild-type and mutant proteins by fXa in complex with factor Va was compared. As shown in Figure 2B, relative to activation of wild-type (Km(app) = 8.9 ± 1.4 and kcat = 390.3 ± 31.5 nM/min/nM), the kcat for the activation of the prethrombin-2 mutant (Km(app) = 5.4 ± 0.5 and kcat = 100.9 ± 3.9 nM/min/nM) was impaired approximately 4-fold whereas the Km(app) was slightly improved. These results suggest that the autolysis loop of the zymogen may contribute approximately 3- to 4-fold to its high rate of activation by fXa in the prothrombinase complex.
Amidolytic activity
Kinetic analysis of the hydrolysis of the chromogenic substrate SpPCa by either wild-type or mutant APC suggested that the kcat for cleavage by the APC mutant (56.9 ± 1.0 s-1) was approximately 2-fold higher than the corresponding value for the wild-type protease (33.2 ± 0.4 s-1). Furthermore, relative to Km for the wild-type protease (134.2 ± 5.3 μM), Km for the mutant (92.7 ± 6.1 μM) was also slightly improved (Table 1). These results are consistent with previous data that elimination of the glycosylation site of the autolysis loop of APC leads to an approximately 2-fold improvement in the amidolytic activity of the mutant protease.30 The improved amidolytic activity of the mutant supports the proposal that the substitution of the autolysis of APC with the corresponding loop of fXa does not adversely affect the folding and reactivity of the catalytic pocket of the mutant protease. Unlike an improvement in the amidolytic activity of the APC mutant, both wild-type and mutant thrombin exhibited similar amidolytic activities toward SpPCa (Table 1). Thus, mutagenesis did not adversely affect the conformation and the reactivity of the catalytic pocket of mutant proteases. In agreement with this proposal, Ki values for the interaction of all mutants with the S1 site-specific competitive inhibitor of serine proteases, p-aminobenzamidine, were comparable or slightly improved (Table 1).
Kinetic constants for the cleavage of Spectrozyme PCa and inhibition by p-aminobenzamidine (PAB) for wild-type and mutant enzymes
Km, μM | kcat, S−1 | kcat/Km, s−1 μM−1 | Ki (PAB), μM | |
|---|---|---|---|---|
| APC | 134.2 ± 5.3 | 33.2 ± 0.4 | 0.25 ± 0.01 | 40.2 ± 2.7 |
| APC-fXa143-154 | 92.7 ± 6.1 | 56.9 ± 1.0 | 0.61 ± 0.05 | 19.3 ± 1.1 |
| Thrombin | 6.5 ± 1.5 | 80.3 ± 5.0 | 12.4 ± 3.6 | 51.6 ± 2.7 |
| Thrombin-fXa143-154 | 7.5 ± 0.8 | 82.2 ± 2.2 | 11.0 ± 1.5 | 51.9 ± 2.8 |
Km, μM | kcat, S−1 | kcat/Km, s−1 μM−1 | Ki (PAB), μM | |
|---|---|---|---|---|
| APC | 134.2 ± 5.3 | 33.2 ± 0.4 | 0.25 ± 0.01 | 40.2 ± 2.7 |
| APC-fXa143-154 | 92.7 ± 6.1 | 56.9 ± 1.0 | 0.61 ± 0.05 | 19.3 ± 1.1 |
| Thrombin | 6.5 ± 1.5 | 80.3 ± 5.0 | 12.4 ± 3.6 | 51.6 ± 2.7 |
| Thrombin-fXa143-154 | 7.5 ± 0.8 | 82.2 ± 2.2 | 11.0 ± 1.5 | 51.9 ± 2.8 |
The kinetic constants were calculated from the cleavage rate of increasing concentrations of SpPCa (0.75-2000 μM) by wild-type and mutant APC (2 nM) and wild-type and mutant thrombin (0.5 nM) in TBS/Ca2+ as described in “Materials and methods.” The kinetic values are averages of 3 measurements ± standard errors. The Ki values for PAB were measured from the decrease in initial rates of S2266 hydrolysis produced by the competitive inhibitor in TBS/Ca2+. The global fitting of data to a competitive binding equation yielded values for Ki (last column) and Km and kcat values for the hydrolysis of S2266 (146.3 ± 8.2 μM and 8.9 ± 0.1 s−1 for APC; 114.4 ± 5.7 μM and 19.0 ± 0.2 s−1 for APC-fXa143-154; 146.5 ± 6.1 μM and 12.8 ± 0.2 s−1 for thrombin; and 209.8 ± 9.5 μM and 12.3 ± 0.2 s−1 for thrombin-fXa143-154).
Inactivation by AT
APC is known to be essentially unreactive with AT, although full-length high-affinity heparins are known to mediate a slow APC-AT reaction by a template mechanism.32 Similar to wild-type APC, the mutant APC reacted very slowly with AT in the absence of a cofactor. The incubation of the APC mutant with a high concentration of AT (10 μM) for several hours was required to estimate a second-order association rate constant (k2) of 3.2 M-1s-1 for the reaction (Table 2). Interestingly, however, the mutant APC reacted efficiently with AT in the presence of pentasaccharide (Figure 3A) with a k2 value of 1.2 × 103 M-1s-1. Thus, the reactivity of the mutant with AT was improved 375-fold. In the case of thrombin, it is known that pentasaccharide minimally affects the reactivity of the protease with AT.6 Similar to data in the literature, we measured an approximately 1.5-fold enhanced reactivity for thrombin with AT in the presence of pentasaccharide (Table 2). However, the reactivity of the thrombin mutant with AT in the presence of pentasaccharide was approximately 37-fold higher than that in the absence of the cofactor (Figure 3B; Table 2). These results suggest that differences in the structure of the amino acid residues of the autolysis loops are responsible for differential reactivity of coagulation proteases with the native and activated conformations of AT.
Second-order rate constants for uncatalyzed and heparin-catalyzed reactions of AT with wild-type and mutant enzymes
kuncat, M−1 s−1 | kH5, M−1 s−1 | kH70, M−1 s−1 | kH5/kuncat, fold | kH70/kH5, fold | |
|---|---|---|---|---|---|
| APC | ND | 3.6 ± 0.6 | (2.0 ± 0.2) × 102 | ND | 56 |
| APC-fXa143-154 | 3.2 ± 1.2 | (1.2 ± 0.1) × 103 | (3.1 ± 0.4) × 105 | 375 | 258 |
| Thrombin | (8.7 ± 0.2) × 103 | (13.4 ± 0.5) × 103 | (1.2 ± 0.1) × 108 | 1.5 | 8955 |
| Thrombin-fXa143-154 | (4.9 ± 0.1) × 103 | (1.8 ± 0.05) × 105 | (1.2 ± 0.1) × 108 | 37 | 667 |
kuncat, M−1 s−1 | kH5, M−1 s−1 | kH70, M−1 s−1 | kH5/kuncat, fold | kH70/kH5, fold | |
|---|---|---|---|---|---|
| APC | ND | 3.6 ± 0.6 | (2.0 ± 0.2) × 102 | ND | 56 |
| APC-fXa143-154 | 3.2 ± 1.2 | (1.2 ± 0.1) × 103 | (3.1 ± 0.4) × 105 | 375 | 258 |
| Thrombin | (8.7 ± 0.2) × 103 | (13.4 ± 0.5) × 103 | (1.2 ± 0.1) × 108 | 1.5 | 8955 |
| Thrombin-fXa143-154 | (4.9 ± 0.1) × 103 | (1.8 ± 0.05) × 105 | (1.2 ± 0.1) × 108 | 37 | 667 |
The uncatalyzed (kuncat) and pentasaccharide (kH5) catalyzed second-order rate constants were determined by the incubation of wild-type and mutant proteases (1-2 nM) with AT (0.025-10 μM) in the absence and presence of saturating concentrations of H5 (1-20 μM) at room temperature in TBS/Ca2+. The full-length heparin (kH70) catalyzed values for both wild-type and mutant APC were determined by the same procedures except that enzymes were incubated with 0.25-5 μM AT and catalytic levels of H70 (0.01-2.5 μM). In the case of thrombin, both wild-type and mutant protease (0.5 nM each) were incubated with 200 nM AT and catalytic levels of heparin (0.5-2.0 nM). The second-order rate constants were calculated from the remaining chromogenic substrate activities as described in “Materials and methods.” The fold conformational effect of H5 was calculated from the ratio of the second-order rate constant in the presence of H5 to the same value in the absence of H5 (kH5/kuncat) as described. 16 The fold template effect of the full-length heparin (H70) was calculated from the ratio of the second- order rate constant in the presence of H70 to the same value in the presence of H5 (kH70/kH5) as described.16
ND indicates not determinable since the incubation of APC with AT (10 μM) for 5 hours resulted in no decline in the chromogenic substrate activity of the enzyme.

Dependence of the pseudo-first-order rate constants (kobs) for the inhibition of APC-fXa143-154 and thrombin-fXa143-154 on the AT concentration in the absence and presence of pentasaccharide (H5). (A) The kobs values for the mutant APC were determined from the time-dependent inhibition of the protease (2 nM) at different concentrations of AT in complex with saturating concentrations of pentasaccharide (1-2 μM) in TBS/Ca2+ at room temperature as described in “Materials and methods.” (B) The same as in panel A, except that the kobs values for the inhibition reaction were determined for the mutant thrombin (1 nM) at different concentrations of AT in either the absence (•) or presence (○) of a saturating concentration of pentasaccharide (1 μM). Solid lines in both panels are the best fitof kinetic data to a linear equation.
Dependence of the pseudo-first-order rate constants (kobs) for the inhibition of APC-fXa143-154 and thrombin-fXa143-154 on the AT concentration in the absence and presence of pentasaccharide (H5). (A) The kobs values for the mutant APC were determined from the time-dependent inhibition of the protease (2 nM) at different concentrations of AT in complex with saturating concentrations of pentasaccharide (1-2 μM) in TBS/Ca2+ at room temperature as described in “Materials and methods.” (B) The same as in panel A, except that the kobs values for the inhibition reaction were determined for the mutant thrombin (1 nM) at different concentrations of AT in either the absence (•) or presence (○) of a saturating concentration of pentasaccharide (1 μM). Solid lines in both panels are the best fitof kinetic data to a linear equation.
Despite the lack of reactivity of AT with APC, we previously demonstrated that an approximately 70-saccharide-long high-affinity heparin catalyzes the reactivity of APC with AT with a k2 of 200 M-1s-1 by a template mechanism at physiologic levels of calcium.32 To evaluate the extent of the reactivity of the APC mutant with AT in the presence of heparin, the inactivation of the mutant protease was monitored in the presence of increasing concentrations of the AT-heparin complex. As shown in Figure 4, kobs values for reaction with both wild-type and mutant APC showed a saturable dependence on the concentration of the AT-heparin complex, indicating the saturation of an intermediate AT-heparin-protease encounter complex prior to formation of a stable, covalent complex as described.32 Nonlinear regression analysis of data by a hyperbolic equation yielded Kd values for the ternary complex dissociation constants and k values for the rate constants of the stable complex formation according to the following scheme:
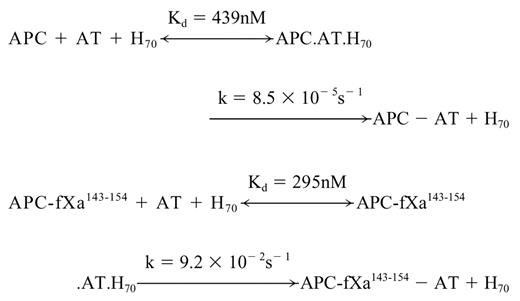

Dependence of the pseudo-first-order rate constants (kobs) for the inhibition of wild-type and mutant APC on the AT-heparin complex concentrations. (A) The kobs values were determined from the time-dependent inhibition of wild-type APC (2 nM) at different concentrations of the AT-heparin (H70) complex (x-axis) in TBS/Ca2+ at room temperature as described in “Materials and methods.” (B) The same as in panel A, except that the kobs values were determined for the APC-fXa143-154 mutant. Solid lines in both panels are best fit of kinetic data to a hyperbolic equation.
Dependence of the pseudo-first-order rate constants (kobs) for the inhibition of wild-type and mutant APC on the AT-heparin complex concentrations. (A) The kobs values were determined from the time-dependent inhibition of wild-type APC (2 nM) at different concentrations of the AT-heparin (H70) complex (x-axis) in TBS/Ca2+ at room temperature as described in “Materials and methods.” (B) The same as in panel A, except that the kobs values were determined for the APC-fXa143-154 mutant. Solid lines in both panels are best fit of kinetic data to a hyperbolic equation.
The k2 values, calculated from the ratio of the rate constants (k) to Kd values for both wild-type (1.9 × 102 M-1s-1) and APC mutant (3.1 × 105 M-1s-1), are in agreement with values determined from the linear slope of subsaturating concentrations of AT (Table 2). The analysis of data in Table 2 suggests that heparin improves the reactivity of the APC mutant with AT by 5 orders of magnitude (kH70/kuncat). Analysis of these data further suggests that the conformational activation of AT by pentasaccharide contributes 375-fold to the rate enhancement (kH5/kuncat), and the template effect of the full-length heparin accelerates the reaction 258-fold (kH70/kH5). It is interesting to note that the catalytic effect of the approximately 70-saccharide heparin in AT inhibition of fXa is also mediated by a combination of approximately 300-fold acceleration through a conformational activation of the serpin and approximately 200- to 300-fold acceleration through a template mechanism at physiologic levels of calcium.7 Moreover, because the reactivity of the mutant thrombin with AT in the presence of pentasaccharide was markedly improved, the ratio of k2 in the presence of H70 to k2 in the presence of H5 (kH70/kH5) was reduced to 667-fold (Table 2), further suggesting that the grafting of the fXa autolysis loop on thrombin has altered the specificity of the mutant protease so that it reacts with the AT-heparin complex by a fXa-like mechanism. These results clearly support our proposal that the autolysis loop of coagulation proteases is critical for their ability to recognize the activated conformation of the serpin.
SDS-PAGE analysis of the APC-AT complex formation
To directly monitor the extent of complex formation of proteases with AT, SDS-PAGE analysis was carried out following incubation of equimolar concentration of enzymes with the serpin in the absence and presence of heparin cofactors. As shown in Figure 1, consistent with the kinetic data, APC did not form a detectable amount of complex with AT in either the absence or presence of pentasaccharide (lanes 4 and 5). However, in the presence of the approximately 70-saccharide-long heparin, a faint high-molecular weight band representing a stable APC-AT complex appeared on the gel (Figure 1 lane 6). On the other hand, while there was no complex formed between the APC mutant and AT in the absence of the cofactors (Figure 1 lane 7), the APC mutant effectively formed a stable complex with the serpin in the presence of heparin cofactors (Figure 1 lanes 8-9). SDS-PAGE analysis also indicated that, similar to wild-type thrombin, the mutant thrombin forms a high-molecular weight stable complex with AT in both the absence and presence of pentasaccharide (data not shown). Moreover, similar to thrombin, the reactivity of the mutant thrombin with the AT-heparin complex in the substrate pathway of the reaction was slightly elevated. In agreement with previous results, an inhibition stoichiometry of approximately 1.5 was observed for both wild-type and mutant thrombins with the AT-heparin complex.33
Discussion
Although trypsin and most coagulation proteases can react with AT, only fIXa and fXa can discriminate between the native and activated conformations of the serpin.6,8-10 Thus, unlike a 300- to 600-fold improved reactivity of fIXa and fXa with AT in the presence of pentasaccharide, no significant difference (< 2-fold) has been observed in the reactivity of trypsin or thrombin with the serpin in the presence of the cofactor.6,34 The molecular basis for the differential reactivity of coagulation proteases with the alternative conformations of AT is not completely understood. Structural data have indicated that the inactive native conformation of the reactive site loop of AT is caused by the preinsertion of 2 N-terminal P14 and P15 (nomenclature of Schechter and Berger35 ) residues of the loop into β-sheet A of the serpin and that the binding of the cofactor to AT causes the expulsion of this inserted region and, thereby, the activation of the serpin.2,36 However, recent mutagenesis data revealed that a pentasaccharide-mediated exposure of a cryptic exosite, outside the reactive site loop of the serpin on strand 3 of β-sheet C, may primarily be responsible for the rate-accelerating effect of the cofactor in both fIXa and fXa inhibition by the activated serpin.13,14 Consistent with the hypothesis that the newly available exosite on AT is an interactive site for a complementary exosite on fIXa and fXa, we demonstrated that the AT reactivity of the Ala substitution mutants of Arg150, a conserved residue in the autolysis loops of both fIXa and fXa, is specifically impaired by an order of magnitude in the presence of pentasaccharide.15,16 There are 2 additional basic residues, Arg143 and Lys147, that are also conserved in both fIXa and fXa; however, mutagenesis of these residues did not change the reactivity of mutant proteases with the activated conformation of AT.15,16 Noting the greater than 300-fold higher reactivity of these proteases with the heparin-activated AT, the interaction of the basic residues of the autolysis loop with the activated conformation of AT did not account for all rate-accelerating effects of pentasaccharide in the inhibition reactions. Thus, to identify other potential interactive sites on fXa, we systematically analyzed the AT reactivity of a series of fXa mutants containing substitutions at essentially all surface loops known to be involved in determining the substrate and inhibitor specificity of coagulation proteases including 39, 60, 70, 90, and the sodium-binding 220 loops.37-40 The results, however, suggested that none of these surface loops is specific for the recognition of the activated serpin, raising the possibility that the autolysis loop of the protease is the only recognition site for the activated serpin; but in addition to basic residues, other residues and/or structural features of the autolysis loop contribute to the recognition mechanism of the activated serpin.
Comparisons of the sequences of the autolysis loops of coagulation proteases from residues 143-154 suggested that this loop is 11 residues long in both fIXa and fXa, but it is longer by 5 to 6 residues in both thrombin and the nontarget protease APC. To test the possibility that structural difference in the autolysis loop of coagulation proteases are primarily responsible for their differential reactivity with the alternative conformation of AT, we prepared mutants of APC and thrombin in which the autolysis loop of both proteases were replaced with the corresponding loop of fXa. As shown, similar to fXa, the reactivity of the APC mutant with AT was improved 375-fold in the presence of pentasaccharide. Similarly, the reactivity of the mutant thrombin with the activated serpin was also markedly improved. Thus, in contrast to an approximately 1.5-fold difference in the reactivity of thrombin with AT in the absence and presence of pentasaccharide, the difference in reactivity was increased to approximately 37-fold for the mutant thrombin. Assuming that the ratio of the rate constant in the presence of pentasaccharide to the same value in the absence of the cofactor represents the fold cofactor effect, the results presented in Table 2 suggest that the reactivity of the mutant thrombin with the active conformation of AT has been improved approximately 37-fold. Alternatively, however, since AT is believed to exist in solution in equilibrium between an inactive loop inserted conformation and an active loop exposed conformation,2 and the relative contribution of each state to the rate constants presented in Table 2 is not known, it may thus be safer to measure the fold rate-accelerating effect of pentasaccharide from the differences in the rate constants between the wild-type and mutant thrombins. In this case, our results would suggest that the mutant thrombin reacts with the activated conformation of AT with approximately 14-fold improved reactivity (ratio of the mutant kH5 to the wild-type kH5; Table 2). We believe that the reason for the lack of a full recovery of the pentasaccharide effect with the thrombin mutant is likely due to the presence of the 60-insertion loop at the entrance of the active-site pocket of thrombin, which is known to restrict the specificity of the protease interaction with macromolecular substrates and inhibitors.37 In support of this hypothesis, we previously showed that the deletion of 4 residues (Tyr, Pro, Pro, and Trp) from this loop results in a mutant (des-Tyr-Pro-Pro-Trp) that also reacts with the heparin-activated AT with 10- to 20-fold improved rate constants.28 Taken together, our results clearly suggest that the autolysis loops of coagulation proteases play a key role in their differential reactivity with the native and activated conformations of the serpin.
In support of the kinetic data, SDS-PAGE analysis of the inhibition reactions demonstrated that, unlike wild-type APC, the mutant APC formed a stable AT-protease complex in the presence but not in the absence of pentasaccharide. We have previously demonstrated that in addition to acceleration by an activation mechanism, an approximately 70-saccharide-long high-affinity heparin promotes the AT inhibition of fXa by approximately 200- to 300-fold through a template mechanism by bridging both the serpin and protease in 1 complex at a physiologic concentration of calcium.7 Since APC also has a basic exosite that interacts with heparin,21 we postulated that the full-length heparin fraction may accelerate the AT inhibition of the APC-fXa143-154 mutant by a fXa-like mechanism. Indeed, as shown in Figure 4, similar to the AT-fXa reaction, a 2-step reaction mechanism for the AT inhibition of the mutant APC was observed in the presence of the full-length heparin. Moreover, similar to inhibition of fXa, the cofactor effect of heparin in AT inhibition of the mutant APC was mediated through a 375-fold acceleration by a conformational activation of the serpin and an approximately 258-fold acceleration through a template mechanism. These results for the first time clearly establish that differences in the structure of the autolysis loops are responsible for the differential reactivity of coagulation proteases with the native and heparin-activated conformations of AT.
Prepublished online as Blood First Edition Paper, June 3, 2004; DOI 10.1182/blood-2004-03-1092.
Supported by grants awarded by the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL 62565 and HL 68571 [A.R.R.]).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
We would like to thank Audrey Rezaie for proofreading of the manuscript.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal