

In this issue of Blood, McReynolds et al1 report a study of extended genetic testing that revealed clinically unrecognized forms of germline aplastic anemia (AA) or inherited bone marrow failure disorder (IBMFD) in patients undergoing hemopoietic cell therapy (HCT). Their cohort of 732 patients with AA underwent either a related or unrelated HCT over nearly 30 years in centers affiliated with the Center for International Blood and Marrow Transplant Research. All patients had a blood sample available for whole-exome analysis.
AA, the quintessential disorder of bone marrow failure, can be acquired or inherited (inherited bone marrow failure disorder [IBMFD]). Acquired forms are usually immune-mediated, whereas inherited forms may be caused by DNA repair defects (Fanconi anemia), short telomere syndromes (dyskeratosis congenita), ribosomopathies (Shwachman-Diamond and Diamond-Blackfan anemia), or other germline mutations (GATA2, RUNX1). Acquired severe AA (SAA) is preferentially treatedwith allogeneic HCT (vs immunosuppressive therapy [IST]) in patients with a suitable donor. Immunosuppression is not a rational therapeutic option in inherited AA, and HCT, when appropriate, requires optimization for the underlying disorder. Thus, knowledge of any germline etiology of the patient's AA has therapeutic relevance.
McReynolds et al used a curated list of 104 genes relevant to hematopoietic differentiation, DNA damage response, telomere, and ribosome biology. After variant selection, they studied the correlation between potential germline mutations and specific post-transplant outcomes. With a rigorous statistical approach, they identified single-nucleotide variants and copy-number variants in 48 patients (6.6%). One-third were adult patients; however, only 3 patients with identified mutations were >40 years of age.
Patients with AA who had genetic evidence of previously unrecognized IBMFD present in the pre-HCT sample experienced worse survival, irrespective of the transplant platform (myeloablative or reduced intensity [RIC] conditioning) or donor relationship. The increased mortality was attributed to organ failure, with the highest rates in those patients with mutations identified in DNA damage response genes followed by telomere biology genes (see figure). These patients unfortunately seem to “melt” with HCT. Patients who were only noted to be carriers of pathogenic variants experienced outcomes similar to presumed acquired cases. In the no-variant group, graft-versus-host disease (GVHD) was the most common cause of death.
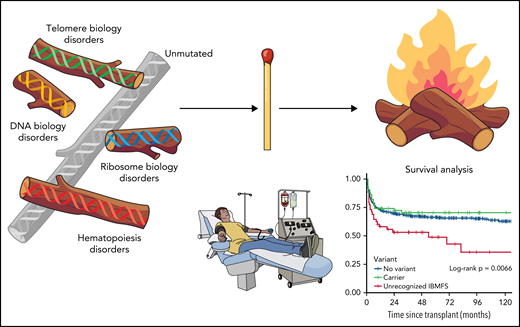
Transpant outcomes for aplastic anemia affected by inherited predisposition. Professional illustration by Patrick Lane, ScEYEnce Studios.
Transpant outcomes for aplastic anemia affected by inherited predisposition. Professional illustration by Patrick Lane, ScEYEnce Studios.
With ever-evolving diagnostic techniques2,3 and increasing recognition of genetic etiologies of AA,4 the standard diagnostic algorithm for AA has become multifaceted. The combination of etiologic heterogeneity with increasingly earlier use of HCT5,6 emphasizes the need for an appropriate diagnostic work to guide the therapeutic paths pursued for all patients with AA.
Over the course of this study, there were lower percentages of unrecognized IBMFD in each subsequent time interval (12 of 236 from 2011 through 2015, compared with 36 of 496 from 1989 through 2010) suggesting increasing awareness of the need to properly diagnose inherited predisposition. This result also coincides with improved use of functional testing, such as peripheral blood lymphocyte telomere length measurement by flow-fluorescence in situ hybridization7 and chromosome breakage with diepoxybutane to rule out short telomere syndromes and Fanconi anemia. McReynolds et al did not have access to fresh plasma to do these tests specifically but were able to confirm the telomere lengths in those mutation-bearing patients by polymerase chain reaction. This series confirms that baseline assessment for these disorders is the minimum mandatory workup before HCT for AA. Furthermore, the study strengthens the case for more comprehensive germline testing such as whole-exome sequencing in all patients with SAA <40 years of age, as 45 of the 48 identified lesions were in this age range.
However, it must be noted that McReynolds et al did not have the ability to do paroxysmal nocturnal hemoglobinuria (PNH) testing on these samples. It is known that the presence of a PNH clone represents a marker of acquired AA disease and can be identified by flow cytometry.8 Genetic testing is costly and not readily accessible in many parts of the world, so the use of PNH clones with the aforementioned functional testing is a cost-conscious initial step. This cohort study would also suggest that adults >40 years of age with negative function testing and a PNH clone may not require additional genetic work-up, especially in resource-limited areas.
It is widely recognized that patients with short telomere syndromes and Fanconi anemia must undergo a less intensive HCT conditioning regimen to avoid morbidity and mortality. McReynolds et al nicely illustrate the lengths required to document these mutations accurately to avoid the "firestorm" of organ toxicity and mortality from HCT. Indeed, even RIC regimens had similarly bad outcomes in this study. Given that 31% of the patients with new mutations documented had variants in telomere biology genes, preparatory regimens that even further attenuate conditioning for these patients are important for future clinical investigations. Lastly, the cause of death in the patients without any identified variants was GVHD, not organ toxicity. There is currently an increased focus on the use of very intensive GVHD regimens in nonmalignant diseases, such as AA,9 to address these potential GVHD complications.
There has been a marked improvement in outcomes of HCT with unrelated donors10 and even those with mismatched donors5 in recent years. In a young patient with a concern for unrecognized IBMFD, there is often concern about higher rates of GVHD with the use of an unrelated donor competing with the worry about the use of a related donor with same mutational issues. Reassuringly, there was not a difference by donor relationship in this cohort, suggesting it is the patient and the conditioning that are critical in HCT optimization for SAA. Interestingly, 5 of 7 patients with unrecognized IBMFD had DNA available from their related donor, and only 1 donor was a carrier. It is more likely that prompt recognition of IBMFD will prompt testing of at-risk relatives who are being considered as a potential donor for related HCT.
The relevance of the data from McReynolds et al to the current rationale for screening to differentiate acquired and inherited AA is very clear. Thorough and comprehensive germline genetic testing for younger patients with AA (outside of those with PNH and 6pCNLOH clones) will inform the patient’s care and allow optimized outcomes. The future of the marrow failure field will be additional demonstration of the potential harm from the use of immunosuppressive therapy in patients with correctly diagnosed IBMFD as well as lack of benefit. Thus, a more personalized and tailored HCT approach will increase survival in all patients with AA and avoid outcomes that go “up in flames.”
Conflict-of-interest disclosure: The author declares no competing financial interests.

