In this issue of Blood, Tanaka et al1 identify a mechanistic link between the high co-occurrence of mutations in the splicing factor SF3B1, with inversion or translocation of chromosome 3 in acute myeloid leukemia (AML).
AML with chromosome 3 inversion (inv(3)) or translocation (t(3;3)) is a distinct disease entity in the current World Health Organization classification. Inv(3)/t(3;3) AML has a dismal prognosis because of low response to conventional chemotherapy and early relapse after bone marrow transplantation.2,3 It is known that inv(3) and t(3;3) chromosome rearrangements cause marked overexpression of the zinc finger transcription factor, EVI1, because of enhancer hijacking of a GATA2 distal enhancer.4 Prior sequencing studies have identified co-mutation of RAS pathway genes and the splicing factor SF3B1 in inv(3)/t(3;3) myeloid malignancies, however the mechanisms by which these additional mutations cooperate with inv(3)/t(3;3) to influence disease severity or susceptibility to AML-directed therapies are not well understood.1,5,6
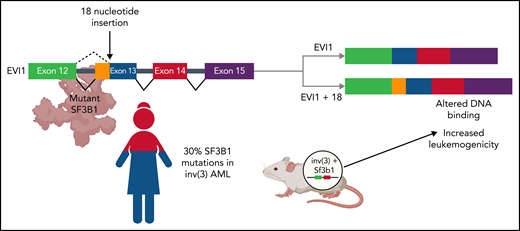
In this study, Tanaka et al assembled a cohort of 109 patients with inv(3)/t(3;3) myeloid malignancies and found that SF3B1 is the most frequently mutated gene in inv(3)/t(3;3) myeloid neoplasms (see figure). This finding is notable, as SF3B1 is the most frequently mutated splicing factor in myelodysplastic syndrome (MDS). In contrast to patients with inv(3)/t(3;3) AML, patients with SF3B1-mutant MDS have a highly favorable prognosis in terms of overall survival and a low risk of transformation to AML.7 Intriguingly, SF3B1 mutations appear to be present in the dominant clone in both inv(3)/t(3;3) AML and SF3B1-mutant MDS, with a variant allele frequency of ∼40% in both diseases. To determine whether SF3B1 mutations affect disease progression in inv(3)/t(3;3) AML, Tanaka et al crossed transgenic mice expressing a humanized inv(3) allele with Sf3b1-mutant mice. They found that humanized inv(3)/Sf3b1 double-mutant hematopoietic stem cells caused earlier lethality compared with humanized inv(3) alone when transplanted into wild-type recipient mice (see figure).

Functional consequences of SF3B1 co-mutation in AML with inv(3) or t(3;3). In a cohort of 109 patients with inv(3)/t(3;3) AML, mutations in the core RNA splicing factor, SF3B1, were identified in >30% of patients (bottom left). Coexpression of humanized inv(3) and mutant Sf3b1 causes increased death from leukemia compared with inv(3) alone in transgenic mice (bottom right). Aberrant mRNA splicing by mutant SF3B1 causes the insertion of 18 nucleotides between exons 12 and 13 of the transcription factor EVI1. This novel isoform, EVI1 + 18, binds to different regions of the genome, leading to the increased expression of genes associated with leukemogenesis (top). Figure created with BioRender.com.
Functional consequences of SF3B1 co-mutation in AML with inv(3) or t(3;3). In a cohort of 109 patients with inv(3)/t(3;3) AML, mutations in the core RNA splicing factor, SF3B1, were identified in >30% of patients (bottom left). Coexpression of humanized inv(3) and mutant Sf3b1 causes increased death from leukemia compared with inv(3) alone in transgenic mice (bottom right). Aberrant mRNA splicing by mutant SF3B1 causes the insertion of 18 nucleotides between exons 12 and 13 of the transcription factor EVI1. This novel isoform, EVI1 + 18, binds to different regions of the genome, leading to the increased expression of genes associated with leukemogenesis (top). Figure created with BioRender.com.
SF3B1 is a core component of the spliceosome that is critical for recognition of the branch point sequence within the introns of pre–messenger RNA (mRNA) and the early stages of splicing catalysis. Mutations in SF3B1 cause the mutant protein to identify an upstream, “cryptic” branch point sequence, leading to the incorporation of intronic nucleotides into a misspliced mRNA transcript. In MDS, up to half of these aberrantly spliced transcripts are predicted to undergo nonsense-mediated mRNA decay (NMD), a quality control mechanism that eliminates misspliced transcripts that contain premature stop codons or frameshifts.8,9 NMD-mediated degradation of aberrantly spliced transcripts from genes involved in heme biosynthesis is thought to partially explain the characteristic anemia seen in patients with SF3B1-mutant MDS.
In the current study, Tanaka et al identify a distinct functional consequence of mutant SF3B1-mediated aberrant splicing in inv(3)/t(3;3) AML. They observed that mutant SF3B1 expression in inv(3)/t(3;3) AML cell lines, patient samples, or humanized inv(3) transgenic murine cells is associated with the use of a cryptic branch point within intron 12 of the EVI1 pre-mRNA. This leads to the generation of a novel isoform, EVI1 + 18, that contains an additional 18 nucleotides between exons 12 and 13 (see figure). This 18-nucleotide insertion leads to a 6-amino-acid in-frame insertion at the C-terminal end of the 10th zinc finger of EVI1. Tanaka et al compared the genomic distribution of EVI1 and EVI1 + 18 in inv(3)/t(3;3) mutant AML cell lines expressing wild-type or mutant SF3B1 and found that EVI1 + 18 bound to a distinct group of genes associated with increased leukemogenesis including MEIS1. Moreover, they identified distinct biological effects associated with EVI1 + 18 expression. When overexpressed in primary murine hematopoietic cells, EVI1 + 18 caused increased proliferation and increased clonogenic capacity compared with canonical EVI1.
The elegant functional studies performed by Tanaka et al suggest that SF3B1 mutations in inv(3)/t(3;3) AML promote an even more aggressive leukemia phenotype caused by EVI1 + 18 mediated upregulation of genes involved in leukemogenesis. Although they did not identify a difference in the overall survival of patients with SF3B1-mutant inv(3)/t(3;3) AML compared to patients with wild-type SF3B1 and inv(3)/t(3;3) AML in their cohort, it is intriguing to consider whether mutant SF3B1 expression may be a therapeutic vulnerability in inv(3)/t(3;3) AML. Previous studies have shown that cells with splicing factor mutations are more sensitive to drugs that interfere with normal spliceosome function. Indisulam is an agent that causes the proteasome-mediated degradation of an essential RNA splicing factor, RMB39.10 Tanaka et al found that the 50% inhibitory concentration for indisulam is lower in SF3B1-mutant inv(3)/t(3;3) AML cell lines than in SF3B1 wild-type inv(3)/t(3;3) AML cell lines. They also used minigene assays to define the nucleotide sequences required for mutant SF3B1-mediated EVI1 mis-splicing.
This study provides an important foundation for the continued development of targeted therapies for aggressive myeloid malignancies. Future studies using the preclinical models developed by Tanaka et al may help evaluate how synthetic introns, introns within a synthetic gene designed to be selectively spliced by mutant but not wild-type SF3B1,11 may be used to selectively induce the expression of toxins or antineoplastic agents in SF3B1-mutant AML cells.
Conflict-of-interest disclosure: The author declares no competing financial interests.