Key Points
A combination with paclitaxel overcomes the resistance of late-stage NHL to macrophage immune checkpoint inhibitors.
Paclitaxel synergizes with CD47 blockade for NHL treatment by inducing a phagocytosis-favorable tumor microenvironment.

Abstract
Tumor-associated macrophages (TAMs) are often the most abundant immune cells in the tumor microenvironment (TME). Strategies targeting TAMs to enable tumor cell killing through cellular phagocytosis have emerged as promising cancer immunotherapy. Although several phagocytosis checkpoints have been identified, the desired efficacy has not yet been achieved by blocking such checkpoints in preclinical models or clinical trials. Here, we showed that late-stage non-Hodgkin lymphoma (NHL) was resistant to therapy targeting phagocytosis checkpoint CD47 due to the compromised capacity of TAMs to phagocytose lymphoma cells. Via a high-throughput screening of the US Food and Drug Administration–approved anticancer small molecule compounds, we identified paclitaxel as a potentiator that promoted the clearance of lymphoma by directly evoking phagocytic capability of macrophages, independently of paclitaxel’s chemotherapeutic cytotoxicity toward NHL cells. A combination with paclitaxel dramatically enhanced the anticancer efficacy of CD47-targeted therapy toward late-stage NHL. Analysis of TME by single-cell RNA sequencing identified paclitaxel-induced TAM populations with an upregulation of genes for tyrosine kinase signaling. The activation of Src family tyrosine kinases signaling in macrophages by paclitaxel promoted phagocytosis against NHL cells. In addition, we identified a role of paclitaxel in modifying the TME by preventing the accumulation of a TAM subpopulation that was only present in late-stage lymphoma resistant to CD47-targeted therapy. Our findings identify a novel and effective strategy for NHL treatment by remodeling TME to enable the tumoricidal roles of TAMs. Furthermore, we characterize TAM subgroups that determine the efficiency of lymphoma phagocytosis in the TME and can be potential therapeutic targets to unleash the antitumor activities of macrophages.
Introduction
Non-Hodgkin lymphoma (NHL) is one of the most common cancers, ranking as the fourth to ninth most leading cause of cancer occurrence worldwide.1 The overall 5-year survival of NHL patients ranges from 25% to 75%.2 Chemotherapy regimens such as cyclophosphamide, doxorubicin, vincristine, and prednisone remain the standard treatment approach for NHL. The majority of B-cell lymphomas, representing 80% to 90% of all NHL cases, are CD20+, and thus, anti-CD20 antibodies such as rituximab are widely used for B-cell lymphoma treatment.3 A combination of rituximab and chemotherapy enables the effective treatment of NHL.4 However, once resistance is developed to these therapies, the prognosis for NHL patients is poor, and overall survival becomes significantly shortened.5-8
Recent exciting progress in cancer immunology has identified therapeutics targeting CD47, an antiphagocytic “don’t eat me” signal, as a promising approach for cancer treatment.9-17 Ligation of CD47 with its receptor signal regulatory protein α (Sirpα) on macrophages transduces a negative signaling cascade within the macrophages to inhibit phagocytosis.18 Such immune evasion mechanisms protect “self” body cells from macrophage immunosurveillance, a process termed programmed cell removal (PrCR), involving macrophage recognition and phagocytosis of target cells.19 Many types of cancer cells, including NHL, exploit this self-protective mechanism to evade PrCR by upregulating CD47. Tumor-associated macrophages (TAMs) are often the most abundant immune components in the tumor microenvironment (TME),20-24 and therefore, restoration of PrCR by blocking the phagocytosis checkpoint CD47-Sirpα represents an attractive strategy for cancer treatment. In a recent trial with relapsed or refractory NHL patients, a combination of CD47-blocking antibody Hu5F9-G4 and rituximab showed great promise without clinically significant safety concerns.8
Despite the promising progress, the clinical efficacy of immunotherapies, including macrophage-based immunotherapy, is often limited to a subset of patients. Effective cancer immunotherapies are urgently needed. Here, we report a novel and effective macrophage-based immunotherapy for NHL treatment. Specifically, through high-throughput screening, we discover that paclitaxel, a chemotherapeutic agent, is able to promote PrCR independently of its direct cytotoxic effect toward malignant cells. Paclitaxel significantly enhances the phagocytic ability of macrophages in the TME through the activation of Src family tyrosine kinases (SFK) signaling, and a combination of paclitaxel and CD47 blockade prevents the accumulation of TAM subgroups, which may contribute to the resistance of late-stage NHL to immunotherapy. Our study identifies new immunotherapeutic strategies for NHL and reveals underlying mechanisms of how TAM populations in the TME react to and determine the efficacy of PrCR-based immunotherapy.
Materials and methods
Mice
BALB/c and RAG2−/−γc−/− BALB/c mice were bred in the Animal Resources Center at City of Hope Comprehensive Cancer Center. BALB/c mice were purchased from The Jackson Laboratory. RAG2−/−γc−/− BALB/c mouse strain was a generous gift from Dr. Irving L. Weissman at Stanford University. All the procedures were approved by the Administrative Panel on Laboratory Animal Care at City of Hope Comprehensive Cancer Center.
Generation of macrophages
Mouse bone marrow–derived macrophages (BMDMs) and human monocyte-derived macrophages were generated as previously described.25
Macrophages from day 6 to day 8 of differentiation were used for this study. Detailed methods are described in the supplemental Materials (available on the Blood Web site).
LB-LTMK
NHL lines and macrophages were used for the luminescence-based long-term macrophage killing (LB-LTMK) assay to examine the effects of macrophage-mediated cancer cell killing. Detailed methods are described in the supplemental Materials.
LB-LTMK screen of 147 anticancer small molecules
The US Food and Drug Administration (FDA)-approved oncology drug library including 147 current approved anticancer small molecules was used to identify potential enhancers of CD47 blockades. Detailed methods are described in the supplemental Materials.
Phagocytosis assay by flow cytometry or microscopy
Phagocytosis rate was evaluated with mouse BMDMs or human peripheral blood monocyte-derived macrophages. Phagocytosis was quantified as the percentage of macrophages that phagocytosed cancer cells. Detailed methods are described in the supplemental Materials.
CRISPR editing
CD47 knockdown cells were generated with clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 system followed by fluorescence-activated cell sorting. Detailed methods are described in the supplemental Materials.
Gene knockdown by siRNA in primary macrophages
The small interfering RNA (siRNA) knockdown of genes in primary BMDMs was performed with siGENOME SMARTpool from Dharmacon in 96-well plates. Detailed methods are described in the supplemental Materials.
Mouse and therapeutics
Raji cells expressing a luciferase-eGFP fusion protein were IV injected into RAG2−/−γc−/− BALB/c mice to establish the systemic NHL disease model. Raji, Daudi, SU-DHL-2, and A20 cells were subcutaneously injected to RAG2−/−γc−/− BALB/c mice or BALB/c mice to develop tumors for evaluating local effects of paclitaxel on aCD47. Either nab-paclitaxel or aCD47 was administered as specified in each figure. Detailed methods are described in the supplemental Materials.
Bioluminescence imaging
Tumor progression was routinely monitored by bioluminescence imaging on a Lago X instrument (Spectral Instruments Imaging). Detailed methods are described in the supplemental Materials.
In vivo depletion of macrophages
Macrophages were depleted in vivo through IV injection of clodronate encapsulated in liposomes. Mice were given either clodronate-liposome or phosphate-buffered saline–liposome (200 μL). Detailed methods are described in the supplemental Materials.
Bulk RNAseq of femur macrophages
RNA of sorted femur macrophages (CD11b+F4/80+) were extracted with an RNA Extraction Kit (Qiagen). The samples were submitted to Novogene Inc for library preparation and subsequent RNA sequencing (RNAseq). Detailed methods are described in the supplemental Materials.
Single-cell RNA sequencing
RAG2−/−γc−/− BALB/c mice were engrafted with Raji cells and treated with control vehicle, aCD47, nab-paclitaxel, or a combination of aCD47 and nab-paclitaxel. CD11b+ myeloid cells were sorted from the bone marrow (BM) of the mice and subjected to subsequent RNA sequencing. Single-cell RNA sequencing reads were quantified by cellranger-5.0.0, aligned to mm10 mouse genome. Detailed methods are described in the supplemental Materials.
Analysis of clinical datasets
For analysis of immune cell composition in NHL patient specimens, the gene expression dataset of a cohort of diffuse large B-cell lymphoma (DLBCL) patient specimens was analyzed using the bioinformatics analytical tool CIBERSORT.26 Detailed methods are described in the supplemental Materials.
Statistics
Statistical analysis was performed with GraphPad Prism (version 8). An unpaired t test was used to detect significant differences between groups; analysis of variance (ANOVA) test or log-linear regression analysis was employed when there were >2 groups to be analyzed. The overall survival of mice or humans was evaluated with the Kaplan-Meier method and log-rank test to compare values between groups. Differences with a value of P < .05 were considered to be statistically significant. All results were expressed as mean plus or minus standard deviation of the mean.
Results
Compromised efficacy of CD47-targeted therapy along with NHL progression
The implementation of a successful macrophage-based therapy relies on the PrCR capacity of TAMs. To address whether TAMs retain their ability to perform PrCR during lymphoma progression, we first evaluated the impact of tumor progression on the efficacy of a CD47-blocking antibody (aCD47) with an in vivo model. The binding affinity of Sirpα from BALB/c mice to human CD47 is comparable to and best mimics that of human Sirpα to human CD47.27,28 Therefore, we used RAG2−/−γc−/− mice (deficient in T, B, and natural killer cells but retain functional macrophages)29 with a BALB/c background for evaluating PrCR of human NHL in in vivo settings. The mice were engrafted with a human NHL line (Raji) via IV injection. Bioluminescence imaging indicated aggressive dissemination of disease to the BM compartment (supplemental Figure 1A), and the mice eventually developed clinical signs of disease progression such as hind-limb paralysis. After engraftment, the mice received the first injection of aCD47 on days 4, 8, or 11, followed by another 4 injections. Complete disease remission was observed in mice treated with aCD47 starting at day 4, whereas the mice receiving the first injection at day 11 showed no reduction of disease (Figure 1A-B), indicating a robust impact of tumor progression on the effectiveness of aCD47. Thus, we deemed day 11 as the onset of late-stage lymphoma in this model when aCD47 demonstrated no effects on the disease. Notably, most cancer cells in BM were bound to aCD47 (supplemental Figure 1B), and abundant TAMs were detected in the TME (supplemental Figure 1C), suggesting that the resistance to aCD47 treatment was not due to impaired antibody binding or insufficient TAM infiltration. Raji cells sorted from the femurs of mice on day 11 displayed similar susceptibility to PrCR when CD47 was blocked as compared with their counterparts cultured in vitro (supplemental Figure 1D-E). In contrast, bone marrow (BM) macrophages sorted from Raji-engrafted mice on day 11 demonstrated a significantly decreased capability for phagocytosing Raji cells as compared with those from the nonengrafted mice (Figure 1C).
![Progression of NHL compromised the efficacy of CD47-blocking antibody. (A) Raji-engrafted mice were treated with aCD47 at different days and tumor burden was recorded on day 22 after engraftment; ***P < .001 (1-way ANOVA test); ns, not significant. (B) CD47-blocking antibody administration (7.5 mg/kg) at different days differentially influenced the survival of NHL-bearing mice. Mice in the day 4 treatment group stayed alive and were euthanized upon completion of the experiment on day 50 (dotted line). **P < .01, ***P < .001 (log-rank [Mantel-Cox] test). (C) Evaluation of the phagocytic ability of BM macrophages from control mice and NHL-bearing mice (day 11 upon engraftment) with microscopy-based phagocytosis assay in the presence of CD47 blocking antibody. **P < .01 (1-way ANOVA test).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/22/10.1182_blood.2021013901/3/m_bloodbld2021013901f1.png?Expires=1766001893&Signature=VBBLbrr1rFdzNpnteu~1uN~qUB0E3QW6LuGisdR~P3i3~UkQvWitxji8n7XabZG09U-prk~G~q9lyzlb0w4J~uCU6lZUeQpEQQIj3FswpOt0aGFJ58yQRbAzPC3Q0WpJPqzOlD1HMmyyDMpjsUI6u-lLPLsgz7XGkaJNaYpDZP4xMEn4uS8vCw-rifhmBoHD1zvafIy36nwAA-G4u8F7mi3Cqk66nayrhDwrHRyLuzYbglI2ZrgIIBLiQoHnILSDHK9TvWhcOcYqy6Ec9V2Ypu8JVgq-eS05MQFR6OSv~m~i27h6XMXaoHkpKk6R89ES1SPH7aKxY6Xf0XPuUveDYA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Progression of NHL compromised the efficacy of CD47-blocking antibody. (A) Raji-engrafted mice were treated with aCD47 at different days and tumor burden was recorded on day 22 after engraftment; ***P < .001 (1-way ANOVA test); ns, not significant. (B) CD47-blocking antibody administration (7.5 mg/kg) at different days differentially influenced the survival of NHL-bearing mice. Mice in the day 4 treatment group stayed alive and were euthanized upon completion of the experiment on day 50 (dotted line). **P < .01, ***P < .001 (log-rank [Mantel-Cox] test). (C) Evaluation of the phagocytic ability of BM macrophages from control mice and NHL-bearing mice (day 11 upon engraftment) with microscopy-based phagocytosis assay in the presence of CD47 blocking antibody. **P < .01 (1-way ANOVA test).
Progression of NHL compromised the efficacy of CD47-blocking antibody. (A) Raji-engrafted mice were treated with aCD47 at different days and tumor burden was recorded on day 22 after engraftment; ***P < .001 (1-way ANOVA test); ns, not significant. (B) CD47-blocking antibody administration (7.5 mg/kg) at different days differentially influenced the survival of NHL-bearing mice. Mice in the day 4 treatment group stayed alive and were euthanized upon completion of the experiment on day 50 (dotted line). **P < .01, ***P < .001 (log-rank [Mantel-Cox] test). (C) Evaluation of the phagocytic ability of BM macrophages from control mice and NHL-bearing mice (day 11 upon engraftment) with microscopy-based phagocytosis assay in the presence of CD47 blocking antibody. **P < .01 (1-way ANOVA test).
Identification of paclitaxel as a potentiator of PrCR
These findings suggested that a protective TME against PrCR was being established during lymphoma progression, conferring resistance to PrCR-based therapy. Therefore, we hypothesized that remodeling the TME by counteracting the PrCR-suppressive mechanisms of NHL cells and/or reenabling the PrCR ability of macrophages would overcome this resistance and restore PrCR. To screen for agents with such functions, we first sought to establish an assay that enables evaluating PrCR in a high-throughput manner. Macrophage-mediated PrCR is typically determined by quantifying macrophage populations phagocytosing cancer cells with flow cytometry- or microscopy-based assays, which can become labor intensive and subject to batch-to-batch variations when performed in a high-throughput manner. To overcome these limitations, we developed a high-throughput suitable LB-LTMK assay to evaluate PrCR-mediated cancer cell killing by quantifying the surviving cancer cells. Specifically, cancer cells transduced with luciferase-eGFP were cocultured with macrophages, and the fluorescence (GFP) and/or luminescence signals from surviving cancer cells were quantified (supplemental Figure 2A). We showed that blocking CD47 or Sirpα induced PrCR and thus led to the reduction of surviving cancer cells in a concentration-dependent manner (supplemental Figure 2B) and that luminescence signals were more sensitive than fluorescent signals (supplemental Figure 2C-D). The accuracy of the LB-LTMK assay in determining phagocytosis rate was consistent with that of the flow cytometry–based method (supplemental Figure 2D-E). These data suggested that the LB-LTMK assay is robust for accurate measurement of PrCR with an efficient experimental pipeline, facilitating the screening of novel chemical or genetic regulators of PrCR.
Thus far, around 150 anticancer small molecule compounds have been approved by the FDA, including chemotherapy or targeted therapy agents.30 Although their roles in inducing immune responses have not been well characterized, the biosafety, metabolism, and tumor penetration ability of these drugs have been fully evaluated.31 With the LB-LTMK assay, 147 current FDA-approved anticancer small molecule compounds were screened using Raji cells and BMDMs (supplemental Table 1; Figure 2A-B; supplemental Figure 2F). Notably, our screen reidentified 2 compounds, imiquimod and azacitidine, which were reported previously to potentiate PrCR.32,33 Among all the compounds tested, paclitaxel, a chemotherapeutic drug, demonstrated the strongest effect in enhancing the efficacy of CD47-blocking agents (Figure 2C). We examined PrCR induced by aCD47 whose effect is a sum of blocking CD47 and eliciting phagocytosis with the Fc domain of the antibody11,34,35 and PrCR induced by knocking down CD47 expression on Raji cells, which solely relies on diminishing CD47-Sirpα interaction. We showed that PrCR was significantly enhanced by paclitaxel in both conditions (Figure 2D-E; supplemental Figure 3A). Consistently, using a human macrophage model derived from peripheral blood monocytes, we showed that the combination of aCD47 with paclitaxel led to significantly enhanced clearance of NHL cells (Figure 2F). To understand the underlying mechanisms of paclitaxel-mediated sensitization to CD47 blockade, we treated NHL cells and macrophages with paclitaxel separately and examined the effects on PrCR (Figure 2G). Paclitaxel pretreatment of macrophages was able to strongly enhance PrCR of Raji cells upon CD47 blockade (Figure 2H). In contrast, pretreatment of Raji cells with the same doses of paclitaxel induced minimal increase in PrCR (Figure 2H). The effect of paclitaxel on promoting CD47 blockade–induced PrCR was then verified with multiple NHL cells (supplemental Figure 3B). Minimal cell death of lymphoma cells was induced by paclitaxel treatment or conditioned media from paclitaxel-treated BMDMs (supplemental Figure 3C-D). In addition, coculture with paclitaxel-treated BMDMs showed no effect on the viability of lymphoma cells (supplemental Figure 3E-F). Furthermore, treatment of BMDMs with paclitaxel significantly enhanced not only the potency but also the maximum capacity of aCD47 in clearing Raji cells (Figure 2I). These findings suggested that, intriguingly, paclitaxel can directly stimulate the PrCR ability of macrophages, independently of its cytotoxicity toward NHL cells. Consistently, paclitaxel treatment demonstrated similar roles in enhancing the effects of rituximab36,37 on eliciting macrophage phagocytosis of multiple NHL cells (supplemental Figure 4A-E). Interestingly, when treated with paclitaxel, macrophage cell surface expression of Sirpα was unaltered, and CRT, the receptor used by macrophages for cancer cell recognition,34,38 remained unchanged (supplemental Figure 5A-B), indicating that paclitaxel promotes the PrCR ability of macrophages through alternative mechanisms. In addition, the expression of receptors for the Fc portion of immunoglobulin G, including FcγRI, FcγRIIB, FcγRIII, and FcγRIV, was not significantly changed when macrophages were treated with paclitaxel (supplemental Figure 5C).
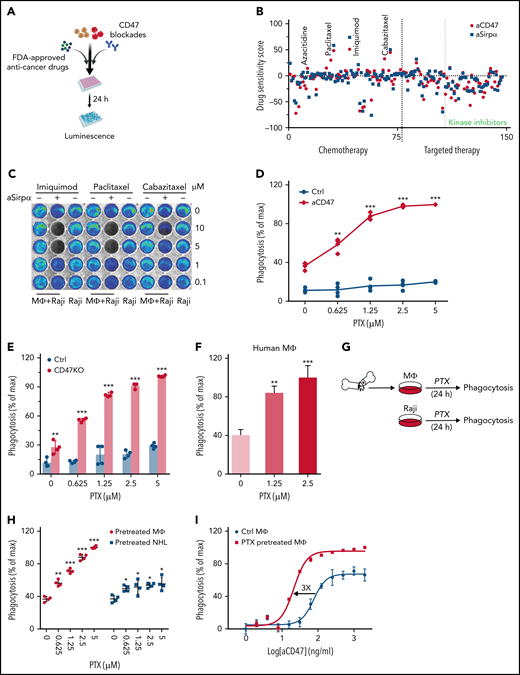
Paclitaxel was identified as an agent potentiating CD47 blockade–mediated phagocytosis through macrophages. (A) A schematic showing the high-throughput screening strategy for identifying agents to enhance CD47 blockade-induced phagocytosis. (B) LB-LTMK high-throughput screens of 147 FDA-approved anticancer small molecule compounds; Raji cells were treated with antibodies blocking CD47-Sirpα interaction (anti-CD47 or anti-Sirpα) and subjected to the LB-LTMK assay; phagocytosis was normalized to dimethyl sulfoxide control; spots represent individual compounds. (C) Representative bioluminescence images of the LB-LTMK assay evaluating the effect of imiquimod, cabazitaxel, and paclitaxel on aSirpα-mediated phagocytosis with Raji cells equipped with luciferase-eGFP as the target cells; BMDMs were used for the assay. (D) Paclitaxel potentiated the effect of CD47 blocking antibody-mediated phagocytosis in a dose-dependent manner. **P < .01; ***P < .001 (1-way ANOVA test). (E) Paclitaxel dose-dependently enhanced phagocytosis of CD47-deficient Raji cells. **P < .01; ***P < .001 (1-way ANOVA test). (F) Paclitaxel enhanced aCD47-mediated phagocytosis of Raji cells by human peripheral blood monocyte-derived macrophages with a luminescence-based phagocytosis assay. **P < .01; ***P < .001 (1-way ANOVA test). (G) A schematic showing the experimental design to evaluate the effect of paclitaxel on BMDMs or Raji cells in enhancing phagocytosis; phagocytosis assays were performed using paclitaxel-treated BMDMs and untreated Raji cells or paclitaxel-treated Raji cells and untreated BMDMs. (H) Pretreatment of BMDMs with paclitaxel significantly enhanced aCD47-mediated phagocytosis of Raji cells. *P < .05; **P < .01; ***P < .001 (1-way ANOVA test). (I) Paclitaxel enhanced the potency of aCD47-mediated phagocytosis of Raji cells by shifting EC50 from 76.48 ng/mL to 21.26 ng/mL and the maximal capacity of aCD47 from clearing 75% of cancer cells within 24 hours to nearly 100% clearance.
Paclitaxel was identified as an agent potentiating CD47 blockade–mediated phagocytosis through macrophages. (A) A schematic showing the high-throughput screening strategy for identifying agents to enhance CD47 blockade-induced phagocytosis. (B) LB-LTMK high-throughput screens of 147 FDA-approved anticancer small molecule compounds; Raji cells were treated with antibodies blocking CD47-Sirpα interaction (anti-CD47 or anti-Sirpα) and subjected to the LB-LTMK assay; phagocytosis was normalized to dimethyl sulfoxide control; spots represent individual compounds. (C) Representative bioluminescence images of the LB-LTMK assay evaluating the effect of imiquimod, cabazitaxel, and paclitaxel on aSirpα-mediated phagocytosis with Raji cells equipped with luciferase-eGFP as the target cells; BMDMs were used for the assay. (D) Paclitaxel potentiated the effect of CD47 blocking antibody-mediated phagocytosis in a dose-dependent manner. **P < .01; ***P < .001 (1-way ANOVA test). (E) Paclitaxel dose-dependently enhanced phagocytosis of CD47-deficient Raji cells. **P < .01; ***P < .001 (1-way ANOVA test). (F) Paclitaxel enhanced aCD47-mediated phagocytosis of Raji cells by human peripheral blood monocyte-derived macrophages with a luminescence-based phagocytosis assay. **P < .01; ***P < .001 (1-way ANOVA test). (G) A schematic showing the experimental design to evaluate the effect of paclitaxel on BMDMs or Raji cells in enhancing phagocytosis; phagocytosis assays were performed using paclitaxel-treated BMDMs and untreated Raji cells or paclitaxel-treated Raji cells and untreated BMDMs. (H) Pretreatment of BMDMs with paclitaxel significantly enhanced aCD47-mediated phagocytosis of Raji cells. *P < .05; **P < .01; ***P < .001 (1-way ANOVA test). (I) Paclitaxel enhanced the potency of aCD47-mediated phagocytosis of Raji cells by shifting EC50 from 76.48 ng/mL to 21.26 ng/mL and the maximal capacity of aCD47 from clearing 75% of cancer cells within 24 hours to nearly 100% clearance.
Resensitization of NHL’s response to aCD47 by paclitaxel
The efficacy of combining aCD47 and paclitaxel was subsequently evaluated in in vivo NHL models. First, we examined the effects of local administration of aCD47 and/or paclitaxel (Figure 3A). Raji, Daudi, or SU-DHL-2 cells were subcutaneously transplanted to RAG2−/−γc−/− mice, and intratumoral treatments of aCD47 and/or paclitaxel were administered. We showed that the treatment with paclitaxel or aCD47 alone slightly inhibited tumor growth, whereas their combination completely abolished tumor growth in the vast majority of mice treated, indicating a synergism between the 2 agents (Figure 3B; supplemental Figure 6A-C). We then examined the efficacy of combining aCD47 and paclitaxel in immunocompetent BALB/c mice engrafted with a mouse NHL cell line (A20). Similarly, we showed that the combination therapy displayed superior antitumor effects than CD47 blockade or paclitaxel alone (Figure 3C). Next, we evaluated the efficacy of the combination therapy in the aforementioned mouse model with advanced systemic NHL disease. Raji cells were IV engrafted into RAG2−/−γc−/− mice followed by the administration of aCD47 at the onset of late-stage lymphoma (day 11 after engraftment) (Figure 3D). We used a nanoparticle albumin-bound (nab) form of paclitaxel for this study, which has been used in clinical applications with improved tumor infiltration and retention and less toxicity.39-42 We showed that nab-paclitaxel demonstrated the same effects in enhancing CD47 blockade–induced PrCR with minimal direct cytotoxicity as paclitaxel (supplemental Figure 6D-F). When a single dose of nab-paclitaxel was IV administered at day 10, 11, or 12, in combination with aCD47, a ∼50-fold reduction of tumor burden was observed, in contrast to 1.3- and threefold reductions of tumor burden, respectively, in mice receiving aCD47 or nab-paclitaxel alone (Figure 3D). In an alternative treatment strategy, a lower dose of nab-paclitaxel, which alone showed no influence on tumor progression, was still able to dramatically enhance the effects of aCD47 in reducing tumor burden (Figure 3E), clearly indicating the role of nab-paclitaxel in promoting the efficacy of CD47-targeted therapy. Furthermore, when BM macrophages were depleted by treating the mice with clodronate-containing liposomes, the effects of aCD47-nab-paclitaxel combination therapy on eliminating NHL cells was abolished (Figure 3F; supplemental Figure 6G-I), highlighting the role of macrophages as the main effectors mediating anticancer effects of the combination therapy.
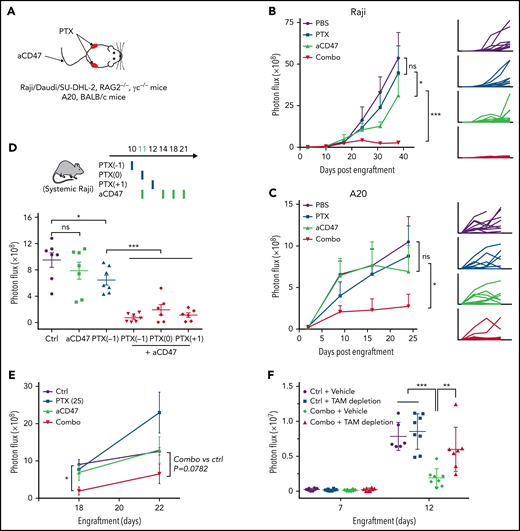
Paclitaxel resensitized the response of aCD47 in NHL-bearing mice. (A) A schematic showing the treatment strategy of the subcutaneous NHL models. Growth of tumors derived from Raji cells in RAG2−/−γc−/− mice (B) and A20 cells in BALB/c mice (C). Mice subcutaneously engrafted with Raji cells or A20 cells were treated with control vehicle, aCD47, paclitaxel, or a combination of aCD47 and paclitaxel. Tumor growth was measured by bioluminescence imaging; *P < .05; ***P < .001 (log-linear regression analysis). (D) A schematic showing the treatment strategy (top); a single dose of nab-paclitaxel (50 mg/kg) largely enhanced the efficacy of aCD47 in mice bearing systemic NHL disease (bottom). *P < .05; ***P < .001 (1-way ANOVA test). (E) A single nontoxic dose of nab-paclitaxel (25 mg/kg) was sufficient to enhance the response to aCD47 in reducing tumor burden. *P < .05 (1-way ANOVA test). (F) Depletion of BM macrophages with clodronate liposome abolished the therapeutic effect of nab-paclitaxel and aCD47 combination. **P < .01, ***P < .001 (1-way ANOVA test). ns, not significant.
Paclitaxel resensitized the response of aCD47 in NHL-bearing mice. (A) A schematic showing the treatment strategy of the subcutaneous NHL models. Growth of tumors derived from Raji cells in RAG2−/−γc−/− mice (B) and A20 cells in BALB/c mice (C). Mice subcutaneously engrafted with Raji cells or A20 cells were treated with control vehicle, aCD47, paclitaxel, or a combination of aCD47 and paclitaxel. Tumor growth was measured by bioluminescence imaging; *P < .05; ***P < .001 (log-linear regression analysis). (D) A schematic showing the treatment strategy (top); a single dose of nab-paclitaxel (50 mg/kg) largely enhanced the efficacy of aCD47 in mice bearing systemic NHL disease (bottom). *P < .05; ***P < .001 (1-way ANOVA test). (E) A single nontoxic dose of nab-paclitaxel (25 mg/kg) was sufficient to enhance the response to aCD47 in reducing tumor burden. *P < .05 (1-way ANOVA test). (F) Depletion of BM macrophages with clodronate liposome abolished the therapeutic effect of nab-paclitaxel and aCD47 combination. **P < .01, ***P < .001 (1-way ANOVA test). ns, not significant.
Clearance of lymphoma by combining paclitaxel and aCD47
To further evaluate the efficacy of this combination therapy, we employed a dosing regimen in which the mice received 1 injection of nab-paclitaxel followed by 2 injections of aCD47 each week for a total of 4 weeks (Figure 4A). Nab-paclitaxel or aCD47 alone was given to mice as a single drug treatment in accordance, with 1 injection of nab-paclitaxel or 2 injections of aCD47 each week. We showed that the combination of nab-paclitaxel and aCD47 stopped lymphoma progression until the mice in other groups reached the endpoint (Figure 4B-C). The strong synergistic effects between the 2 agents were further demonstrated by the significantly prolonged survival of the mice in the combination treatment group (Figure 4D). Importantly, the combination treatment did not influence the body weights of the mice, implying a minor or absence of additional toxicity (Figure 4E). Taken together, we validated that nab-paclitaxel strongly sensitized the response of NHL to CD47 blockades, and the combination strategy yielded superior anticancer effects even in late-stage NHL.

Paclitaxel and aCD47 synergized to yield remarkable antitumor activity in mice bearing systemic NHL disease. (A) A schematic showing the treatment strategy of the systemic Raji models. Mice were treated with an injection of nab-paclitaxel (50 mg/kg) followed by 2 injections of aCD47 (7.5 mg/kg) each week for a total of 4 weeks. (B) Representative animal images showing the efficacy of the combination therapy in reducing tumor burden. (C) Growth of tumors derived from Raji cells through IV injection into RAG2−/−γc−/− mice. Tumor signals were determined by bioluminescence imaging; a combination of nab-paclitaxel and aCD47 stopped the disease progression when other groups of mice reached the endpoint. ***P < .001 (1-way ANOVA test). (D) Survival analysis of animals in panels B and C showing the combination therapy significantly prolonged the survival of the mice. ***P < .001 (log-rank (Mantel-Cox) test). (E) Body weight analysis of animals in panels B and C showing the combination therapy was well tolerated by the mice.
Paclitaxel and aCD47 synergized to yield remarkable antitumor activity in mice bearing systemic NHL disease. (A) A schematic showing the treatment strategy of the systemic Raji models. Mice were treated with an injection of nab-paclitaxel (50 mg/kg) followed by 2 injections of aCD47 (7.5 mg/kg) each week for a total of 4 weeks. (B) Representative animal images showing the efficacy of the combination therapy in reducing tumor burden. (C) Growth of tumors derived from Raji cells through IV injection into RAG2−/−γc−/− mice. Tumor signals were determined by bioluminescence imaging; a combination of nab-paclitaxel and aCD47 stopped the disease progression when other groups of mice reached the endpoint. ***P < .001 (1-way ANOVA test). (D) Survival analysis of animals in panels B and C showing the combination therapy significantly prolonged the survival of the mice. ***P < .001 (log-rank (Mantel-Cox) test). (E) Body weight analysis of animals in panels B and C showing the combination therapy was well tolerated by the mice.
Induction of phagocytosis-capable TAM population by paclitaxel
Next, to understand the molecular mechanisms of how PrCR ability of TAMs is modified during NHL progression and/or in response to paclitaxel treatment, we characterized the molecular phenotype of TAMs in NHL. BM macrophages collected from the femurs of unengrafted mice (day 0) and Raji-engrafted mice at early- (day 4) or late-stage (day 18) lymphoma were subjected to RNA sequencing. A highly similar gene expression profiling was observed between day 0 and day 4 macrophages (Figure 5A). When comparing day 4 and day 18 TAMs, we found 391 genes significantly upregulated and 14 genes significantly downregulated in late-stage lymphoma (Figure 5B; supplemental Figure 7A), indicating a significant change in TAM phenotypes alongside NHL progression and upregulation of most of the altered genes. Interestingly, both M1- and M2-like macrophage markers were upregulated in day 18 TAMs (Figure 5C), suggesting no significant shift of TAMs in late-stage NHL toward either M1- or M2-like macrophages. TAMs are diverse and comprise multiple subpopulations with distinct phenotypes and functions,23 and bulk RNAseq is not able to definitively distinguish between these TAM subgroups. Therefore, we performed single-cell RNA sequencing (scRNAseq) on myeloid cells to understand how nab-paclitaxel treatment remodeled the TME of NHL to induce PrCR. Mice engrafted with Raji cells were treated with control vehicle, nab-paclitaxel, aCD47, or a combination of nab-paclitaxel and aCD47. Treatment started at day 11 after engraftment, and CD11b+ myeloid cells were sorted from the BM and subjected for scRNAseq at day 18. Unbiased analyses were performed on the unified cells, leading to the identification of 20 distinct clusters (supplemental Figure 7B-C). Fifteen to thirty percent of these cells were TAMs, characterized by the expression of macrophage markers F4/80, CD68, and CSF1R, and most of the clusters significantly enriched or depleted upon nab-paclitaxel or combination treatment were TAMs (supplemental Figure 7B-D). We then focused on TAM populations and performed a more defined clustering, leading to the identification of 9 TAM clusters with distinct transcriptomes (Figure 5D-E). We showed that 1 of the clusters (C2) was significantly enriched in TAMs from nab-paclitaxel- and combo-treated mice (Figure 5D-E). MHC II genes (H2-eb1, etc) were significantly upregulated in C2 (Figure 5F). In addition, genes including Pf4, CD209a, Mrc1, Clec10a, and Axl were found to be upregulated and specifically expressed in C2 (Figure 5F-G). Taken together, the identification of these genes allowed us to define C2 as a TAM subgroup induced by nab-paclitaxel treatment. These C2-specific markers were identified in previous studies as important signaling molecules functioning through the activation of protein tyrosine kinases.43-47 For example, the chemokine CXCL4 (PF4) induces Src/Syk and MAPK signaling in neutrophils and monocytes47; phosphorylation of tyrosine residues in AXL mediates intracellular signaling of MEK, phosphatidylinositol 3-kinase (PI3K), and SFK pathways48; and Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN, CD209) activates Ras proteins, leading to the activation of p21-activated kinases, Src kinases, or MEK pathways.49 Consistently, protein kinases were reported to play a role in innate immunity,50,51 and we showed that SFK, BTK, MEK, and PI3K pathways were activated in macrophages during PrCR (Figure 5H). Therefore, we performed an LB-LTMK screen of kinase inhibitors targeting these pathways in macrophages to examine whether these signaling pathways were involved in regulating paclitaxel-mediated enhancement of PrCR. We found that inhibitors targeting PI3K showed no effect on PrCR, whereas BTK and MEK inhibitors nonspecifically diminished PrCR in the presence or absence of paclitaxel (Figure 5I). In contrast, dasatinib and ponatinib (SFK/Abl inhibitors) showed no effects on PrCR by control macrophages but were able to reverse paclitaxel-induced augmentation of PrCR to the basal level (Figure 5I). siRNA-mediated knockdown of Src, but not Abl, lessened PrCR of Raji cells in the presence of aCD47 (supplemental Figure 8A). Imatinib, a specific inhibitor for Abl, showed no such effect (Figure 5I), whereas the more specific SFK inhibitor PP1 significantly blunted paclitaxel-stimulated increase of PrCR (supplemental Figure 8B). In addition, treatment of human THP1 macrophages or mouse BMDMs with paclitaxel induced an enhanced SFK phosphorylation (Figure 5J). Taken together, these results suggested a potential role of paclitaxel in the recruitment and activation of TAM subgroups with enhanced tyrosine kinase signaling to promote PrCR.
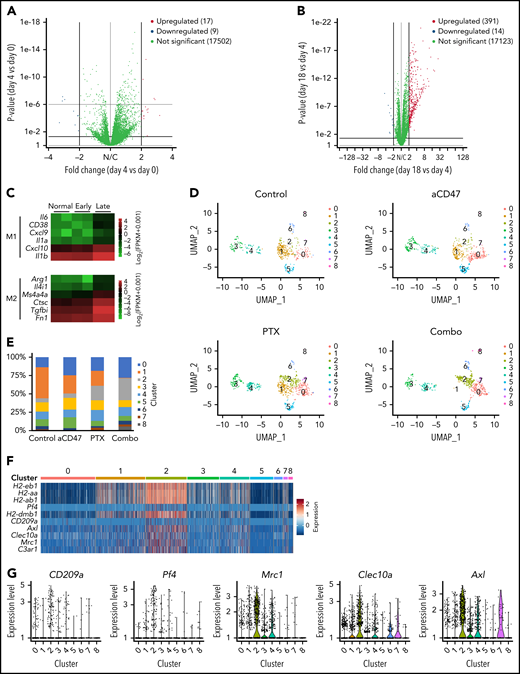

Paclitaxel induced phagocytosis-capable TAM populations. (A-B) Volcano plots showing gene expression of BM macrophages from Raji-engrafted RAG2−/−γc−/− mice at day 0 (normal), day 4 (early), or day 18 (late) of engraftment. Panels A and B showed the comparison of differential gene expression of day 4 vs day 0 (A) and day 18 vs day 4 (B). (C) Heatmap of M1- and M2-like macrophage marker expression in BM macrophages from Raji-engrafted RAG2−/−γc−/− mice at day 0 (normal), day 4 (early) or day 18 (late) of engraftment. (D) UMAP plots of scRNAseq of BM macrophages from Raji-engrafted RAG2−/−γc−/− mice treated with control vehicle, aCD47, nab-paclitaxel, or a combination of aCD47 and nab-paclitaxel. (E) Percentages of cells in clusters 0 to 8 analyzed in panel D. (F) Heatmap of representative genes specifically expressed in cluster C2 from scRNAseq clustered in panel D. (G) Expression of genes Pf4, CD209a, Mrc1, Clec10a, and Axl from scRNAseq clustered in panel D. (H) Immunoblots showing the phosphorylation of protein kinases SFK, BTK, MEK, and PI3K in BMDMs during phagocytosis of Raji cells induced by aCD47. (I) A phagocytosis assay with BMDMs and Raji cells; CD47-blocking antibody was given to all conditions; BMDMs were treated with kinase inhibitors as indicated in the figure. ***P < .001 (1-way ANOVA test). (J) Immunoblots showing Src phosphorylation induced by paclitaxel in human THP1 macrophages and mouse BMDMs. UMAP, Uniform Manifold Approximation and Projection.
Paclitaxel induced phagocytosis-capable TAM populations. (A-B) Volcano plots showing gene expression of BM macrophages from Raji-engrafted RAG2−/−γc−/− mice at day 0 (normal), day 4 (early), or day 18 (late) of engraftment. Panels A and B showed the comparison of differential gene expression of day 4 vs day 0 (A) and day 18 vs day 4 (B). (C) Heatmap of M1- and M2-like macrophage marker expression in BM macrophages from Raji-engrafted RAG2−/−γc−/− mice at day 0 (normal), day 4 (early) or day 18 (late) of engraftment. (D) UMAP plots of scRNAseq of BM macrophages from Raji-engrafted RAG2−/−γc−/− mice treated with control vehicle, aCD47, nab-paclitaxel, or a combination of aCD47 and nab-paclitaxel. (E) Percentages of cells in clusters 0 to 8 analyzed in panel D. (F) Heatmap of representative genes specifically expressed in cluster C2 from scRNAseq clustered in panel D. (G) Expression of genes Pf4, CD209a, Mrc1, Clec10a, and Axl from scRNAseq clustered in panel D. (H) Immunoblots showing the phosphorylation of protein kinases SFK, BTK, MEK, and PI3K in BMDMs during phagocytosis of Raji cells induced by aCD47. (I) A phagocytosis assay with BMDMs and Raji cells; CD47-blocking antibody was given to all conditions; BMDMs were treated with kinase inhibitors as indicated in the figure. ***P < .001 (1-way ANOVA test). (J) Immunoblots showing Src phosphorylation induced by paclitaxel in human THP1 macrophages and mouse BMDMs. UMAP, Uniform Manifold Approximation and Projection.
Prevention of the accumulation of TAM population specific to late-stage NHL by a combination of paclitaxel and aCD47
A TAM cluster (C1) was found not present in mice that received combo-treatment (Figure 5D-E). Importantly, the gene signature of C1 highly overlapped with that of TAMs from late-stage NHL (Figure 6A-B), suggesting that paclitaxel-aCD47 combination remodeled the TME of NHL by inhibiting the accumulation of TAM populations that were favorable for lymphoma progression. Specifically and significantly upregulated genes in C1 include: (1) Socs3, a regulator of cytokine and hormone signaling,52 (2) Sdc4, a syndecan involved in PKC and FGF signaling for cell migration and adhesion,53 (3) Mmp14, a matrix metallopeptidase involved in extracellular matrix remodeling,54 and (4) Arg2, an arginase for arginine hydrolysis55 (Figure 6C). To further evaluate the role of C1 in NHL progression, we performed a supervised analysis of the scRNAseq results based on the aforementioned gene expression profiling of BM macrophages from unengrafted, early-stage NHL and late-stage NHL mice. We identified 5 unique clusters and reidentified a TAM cluster (C1’) that was not present in the tumors of mice receiving combination treatment (Figure 6D-E), and the gene expression profiling of C1’ recapitulated the gene signature of C1 and late-stage NHL (Figure 6F). Lastly, we evaluated this finding by analyzing NHL patient specimens. The gene expression profiling of a cohort of 1078 DLBCL patient specimens at different stages56,57 was analyzed using CIBERSORT.26 Deconvolution of the immune cell populations showed that the immune cell composition in these samples remained largely unaltered across the stage 1 through 4 (supplemental Figure 9A). Consistent with our finding, further analysis examining gene expression in different immune cell populations demonstrated that the genes specifically expressed in cluster C1, Sdc4, Mmp14, and Socs3 were expressed at low or undetectable levels in TAMs of early-stage DLBCL but largely upregulated in TAMs of late-stage specimens (Figure 6G; supplemental Figure 9B). In contrast, a similar gene expression pattern was not observed in other groups of immune cells in the TME, such as T, B, natural killer, and Mast cells (supplemental Figure 9C-K).

A combination of paclitaxel and aCD47 remodeled TME of lymphoma by preventing the accumulation of TAM population specifically present in late-stage NHL. (A) Heatmap of representative genes specifically expressed in cluster C1 from scRNAseq clustered in Figure 5D. (B) Heatmap of genes in panel A in BM macrophages from Raji-engrafted RAG2−/−γc−/− mice at day 0 (normal), day 4 (early), or day 18 (late) of engraftment, analyzed by RNAseq. (C) Expression of genes Socs3, Sdc4, Mmp14, and Arg2 from scRNAseq clustered in Figure 5D. (D) UMAP plots of scRNAseq of BM macrophages from Raji-engrafted RAG2−/−γc−/− mice treated with control vehicle, aCD47, nab-paclitaxel, or a combination of aCD47 and nab-paclitaxel, analyzed by supervised clustering. (E) Percentages of cells in clusters 0 to 4 analyzed in panel D. (F) Heatmap of genes in panel A from scRNAseq clustered in panel D. (G) Expression of genes Socs3, Sdc4, and Mmp14 in macrophages of a cohort of stage 1 to 4 DLBCL patient biopsies, inferred by CIBERSORT; for biopsies with stages 1, 2, 3, and 4, n = 174, 213, 262, and 429. UMAP, Uniform Manifold Approximation and Projection.
A combination of paclitaxel and aCD47 remodeled TME of lymphoma by preventing the accumulation of TAM population specifically present in late-stage NHL. (A) Heatmap of representative genes specifically expressed in cluster C1 from scRNAseq clustered in Figure 5D. (B) Heatmap of genes in panel A in BM macrophages from Raji-engrafted RAG2−/−γc−/− mice at day 0 (normal), day 4 (early), or day 18 (late) of engraftment, analyzed by RNAseq. (C) Expression of genes Socs3, Sdc4, Mmp14, and Arg2 from scRNAseq clustered in Figure 5D. (D) UMAP plots of scRNAseq of BM macrophages from Raji-engrafted RAG2−/−γc−/− mice treated with control vehicle, aCD47, nab-paclitaxel, or a combination of aCD47 and nab-paclitaxel, analyzed by supervised clustering. (E) Percentages of cells in clusters 0 to 4 analyzed in panel D. (F) Heatmap of genes in panel A from scRNAseq clustered in panel D. (G) Expression of genes Socs3, Sdc4, and Mmp14 in macrophages of a cohort of stage 1 to 4 DLBCL patient biopsies, inferred by CIBERSORT; for biopsies with stages 1, 2, 3, and 4, n = 174, 213, 262, and 429. UMAP, Uniform Manifold Approximation and Projection.
Discussion
Here, we discovered that CD47-targeted therapy was highly effective for early-stage NHL, but its efficacy significantly dropped as the disease progressed. NHL cells representing different disease stages demonstrated comparable susceptibility to in vitro CD47 blockade–induced phagocytosis but showed distinct responses to therapies designed to activate PrCR in in vivo settings. CD47-blocking antibodies alone were not able to efficiently induce anticancer responses in late-stage NHL despite the NHL cells being sufficiently bound by the antibodies. Given this discrepancy, even further optimization of the efficacy of CD47 blockade or the administration strategies of blocking agents may not be sufficient to elicit drastic improvement on the efficacy of PrCR therapy for NHL.
Our data suggested that the resistance of late-stage NHL cells to PrCR-based therapy was mainly attributable to the compromised PrCR ability of macrophages in the TME of NHL. The engraftment of Raji cells in mice induced a phenotypic shift of BM macrophages toward a state linked to weaker PrCR capability toward NHL cells. In order to generate potent macrophage PrCR–based cancer immunotherapy, we sought to combine the restoration of PrCR-capable states of macrophages with the blockade of macrophage immune checkpoints. We demonstrated an attractive strategy of repurposing paclitaxel, a chemotherapeutic drug, to be a robust adjuvant to CD47-targeted therapy. We found there to be minimal effect on the viability of NHL cells following paclitaxel treatment, suggesting an anticancer role of paclitaxel independent of its direct cytotoxicity toward NHL cells. In addition to promoting PrCR induced by CD47 blockade, paclitaxel-activated macrophages with enhanced phagocytic ability may elicit stronger phagocytosis toward lymphoma cells through antibody Fc domain–mediated prophagocytic mechanisms and thus may be applied in combinatorial therapy with rituximab and other therapeutic antibodies for NHL treatment. Intriguingly, we showed that paclitaxel reenabled PrCR by not only directly stimulating the phagocytic capacity of BM macrophages but also reversing the phagocytosis-inhibitory TME through the suppression of TAM populations directly linked to NHL progression. A combination with paclitaxel thus significantly amplified the anticancer response of CD47-targeted therapy.
In locations such as the TME, TAMs are often highly heterogeneous and are composed of multiple populations with different or sometimes opposite phenotypes and functions that cannot be precisely defined by the broad classification of macrophages into being either classically activated (M1) or alternatively activated (M2).23,24 A better characterization of TAM subgroups should shed light on the understanding of TAM differentiation and cancer cell–TME interaction. Using single-cell RNA sequencing, we revealed a group of cell surface receptors and secreted factors as novel markers characterizing TAMs induced by nab-paclitaxel, as well as a group of novel markers defining a TAM group that was only present in late-stage NHL whose accumulation was suppressed by the combination PrCR therapy. Our discovery suggested that when used at low doses, without directly inducing cancer cell cytotoxicity, paclitaxel activated SFK signaling in macrophages and promoted the remodeling of the immunosuppressive TME by evoking PrCR-capable TAMs. This finding unveils a new approach for the discovery and implementation of immunotherapies and reveals that when properly induced, SFK signaling can boost the efficacy of cancer immunotherapy. Further mechanistic studies are needed to precisely define the phenotypes of the TAM subgroups, understand their recruitment and polarization states during the treatment with PrCR-based therapies, and investigate their function in facilitating or inhibiting PrCR and/or modifying the TME.
In summary, this study identifies an immunotherapeutic strategy that would lead to a new paradigm of using paclitaxel to activate TAMs and in combination therapy regimens with macrophage-immune checkpoint inhibitors to improve the treatment efficacy for NHL. Because paclitaxel is already a widely used antineoplastic drug, this study will potentially lead to a fast bench-to-bedside translation. The superior efficacy of such a combinatory strategy and the novel roles of TAM subgroups identified and characterized in our study may stimulate further studies for identifying additional agents and therapies targeting macrophages in the TME to enhance their anticancer activity, which may lead to the development of enhanced macrophage-based cancer immunotherapies.
Acknowledgments
The authors thank the excellent technical support of Core Facilities at City of Hope, including the Analytical Cytometry, Animal Resource Center, Small Animal Imaging, Integrative Genomics, and Light Microscopy Core Facilities supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. They thank S. Chen, S. Muend, S. Lai, A. Li, and R. Pillai for helpful discussions, proofreading, or technical assistance. The visual abstract was created with Biorender.com.
This work was supported by the Lymphoma Research Foundation Postdoctoral Fellowship (X.C.), the National Cancer Institute of the National Institutes of Health R00CA201075 (M.F.) and R01CA255250 (M.F.), the Damon Runyon-Dale F. Frey Award for Breakthrough Scientists DFS-22-16 (M.F.), the Margaret E. Early Medical Research Trust Grant (M.F.), the V Foundation for Cancer Research V Scholar Award (V2018-012) (M.F.), and the startup research funding from City of Hope (M.F.). Funds from anonymous donors helped accelerate these studies.
Authorship
Contribution: X.C., Y.W., S.T.R., and M.F. conceived the project; X.C., Y.W., and M.F. designed and performed experiments and analyzed the data; W.Z., X.Z., J.D., and J.W. performed experiments; E.G.G., A.L.E., C.Q., and Z.S. contributed to experimental design and scientific input; and X.C., Y.W., S.T.R., and M.F. wrote and edited the manuscript.
Conflict-of-interest disclosure: M.F. reports a patent pertaining to stimulating TLR/BTK signaling to promote CRT in macrophages assigned to Stanford University; equity and/or consulting with Forty Seven, Inc; and consulting with Innovent Biologics, Inc. The remaining authors declare no competing financial interests.
Correspondence: Steven T. Rosen, Beckman Research Institute, City of Hope, 1500 E Duarte Rd, Duarte, CA 91010; e-mail: srosen@coh.org; and Mingye Feng, Beckman Research Institute, City of Hope, 1500 E Duarte Rd, Duarte, CA 91010; e-mail: mfeng@coh.org.
The sequencing analysis of femur bone marrow macrophages was deposited into the Gene Expression Omnibus (GEO) with accession number GSE190598.
Requests for data sharing may be submitted to Steven T. Rosen and Mingye Feng (srosen@coh.org and mfeng@coh.org).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.