

TO THE EDITOR:
Sickle cell disease (SCD) is a mature erythrocyte disorder that is characterized by severe complications that affect multiple organs.1,2 The cause of this clinical heterogeneity is only partly explained. Clonal hematopoiesis (CH) is an age-related expansion of hematopoietic stem cells (HSCs) that typically carry a somatic mutation in genes associated with hematologic malignancies.3,4 CH has been linked to a higher risk for overt hematological malignancy, coronary heart disease, and all-cause mortality.3-6 It is unknown whether CH modifies the severity of SCD.
Another fundamental question is whether SCD affects the development of CH. Although earlier and/or more frequent CH has been reported in several other inherited hematological disorders,7-9 our current understanding of SCD pathophysiology would not implicate a cell-intrinsic effect on HSCs. However, indirect observations suggest that SCD may have some influence on CH. Patients with SCD have a higher risk for acute leukemia.10-12 Furthermore, a recent report described 2 SCD patients with early graft failure after hematopoietic cell transplantation (HCT) who subsequently developed TP53-mutated therapy-related myeloid neoplasms.13 In both cases, the TP53 mutations were detectable in the recipient prior to transplantation. Finally, autologous cellular therapies based on HSC gene transfer and gene editing or allogeneic HCT with reduced-intensity conditioning are under investigation in SCD.14,15 Given that the consequences of CH-carrying clone expansion are unknown in these settings, it is of paramount importance to determine whether SCD influences CH.
To characterize CH in SCD, we searched for somatic variants in whole-exome sequencing (WES) data from 1459 patients with SCD from 5 distinct cohorts: the Cooperative Study of Sickle Cell Disease (n = 246),16 Genetic Modifier (GEN-MOD, n = 406),17 Differential Response to Hydroxyurea and Incidence of Stroke in Sickle Cell Disease (CIP, n = 456), Mondor/Lyon (n = 322), and BioMe (n = 29). Mean age (± standard deviation) at blood sampling was 23.6 ± 14.0 years (supplemental Figure 1 available on the Blood Web site). SCD subtypes included SS or S/β0 thalassemia (n = 1439) and SC (n = 20). We used the 6848 self-declared African Americans without SCD from the BioMe cohort as controls (including 517 with sickle cell trait [SCT] among the 6227 patients with available genotype).18 Mean age at blood sampling was 50.1 ± 15.1 years (supplemental Figure 2).
We followed the Exome Aggregation Consortium protocol built around the genome aggregation database toolkit best practice.19 We restricted variant discovery to an in-house list of 46 genes that are frequently mutated in CH (supplemental Table 1). We removed all potential germline variants with a variant allele frequency (VAF) ≥ 40% or present in databases (regardless of the minor allele frequency). When remaining DNA was available, we validated each somatic variant detected in patients with SCD using amplicon-based high-depth sequencing (supplemental Figure 3). The complete pipeline and statistical analysis plan are detailed in supplemental Methods.
We identified 15 CH mutations in 15 patients with SCD from 4 distinct cohorts (Table 1) and 199 CH mutations in 189 controls (supplemental Table 2). All of the patients with SCD had SS or S/β0 thalassemia subtype, and 5 were taking hydroxyurea before blood sampling. We found 13 single nucleotide variants (the most common was the age-related C>T transition; n = 6)20 and 2 deletions in patients with SCD. Variants were annotated as missense (n = 10), non-sense (n = 2), frameshift (n = 2), or essential splice site (n = 1), and VAF based on the WES data ranged from 0.025 to 0.26 (median, 0.07). We found CH variants in DNMT3A (n = 10), TP53 (n = 2), TET2 (n = 1), ETV6 (n = 1), and IKZF1 (n = 1).
Individuals with CH were older than those without CH within the SCD and non-SCD cohorts (Figure 1A; supplemental Figure 4A). We also found that CH occurs earlier in patients with SCD compared with non-SCD controls (Figure 1B). The youngest individuals with CH in the SCD and non-SCD groups were 17 and 34 years old, respectively. Furthermore, CH was more prevalent in patients with SCD: in each decade, the proportion of individuals with CH mutations was higher among SCD patients (Figure 1B). In a logistic regression model adjusted for age, sex, and exon capture methodology, SCD status was independently associated with an increase in CH occurrence (odds ratio, 13.5; 95% confidence interval, 3.1-41.9; P = 5.3 × 10−5). The estimated cumulative incidence of CH was higher in patients with SCD compared with controls without SCD (P < .0001, log-rank test). At the age of 50 years, the probability of detecting a CH mutation was 7.1% in patients with SCD and 0.7% in controls (supplemental Figure 5). Although the sequencing depth of targeted genes was higher for the SCD vs the non-SCD WES data sets (42× vs 23×; P < .0001, Student t test), we performed several sensitivity analyses to determine that this difference in sequencing depth does not explain, on its own, our observation of earlier CH in patients with SCD (supplemental Data; supplemental Figures 6-9). Furthermore, to circumvent the sequencing depth heterogeneity among cohorts, we analyzed the BioMe cohort alone because it was processed with the same sequencing pipeline and included 29 patients with SCD. In these BioMe-only analyses, we found that SCD status was still associated with an increase in the occurrence of CH (P = 5.0 × 10−5; supplemental Data). SCT was not associated with earlier occurence of CH (supplemental Figure 10).
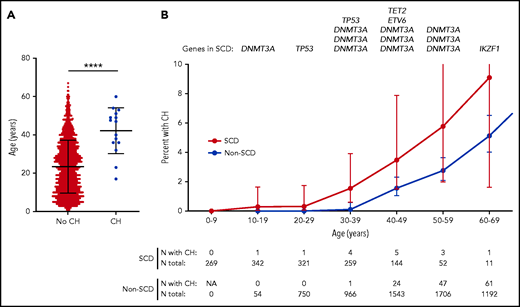
Age distribution of SCD patients with CH. (A) Age distribution of patients with SCD, with and without CH. The mean age ± standard deviation of patients with SCD with and without CH was 42 ± 12 years and 23 ± 14 years, respectively. ****P < .0001, Student t test. (B) Prevalence of CH according to age with 95% confidence interval. Patients with SCD and African American controls without SCD are grouped by decade. The prevalence of CH in controls without SCD who were older than 70 years of age is shown in supplemental Figure 4B. Genes carrying the CH mutation in patients with SCD are shown for each age group. In the groups aged 20 to 29, 30 to 39, 40 to 49, 50 to 59, and 60 to 69 years, the percentage of patients with SCD and CH is 0.3%, 1.5%, 3.5%, 5.8%, and 9.1%, respectively, whereas the percentage of controls with CH is 0%, 0.1%, 1.6%, 2.8%, and 5.1%, respectively. N, number; NA, not applicable.
Age distribution of SCD patients with CH. (A) Age distribution of patients with SCD, with and without CH. The mean age ± standard deviation of patients with SCD with and without CH was 42 ± 12 years and 23 ± 14 years, respectively. ****P < .0001, Student t test. (B) Prevalence of CH according to age with 95% confidence interval. Patients with SCD and African American controls without SCD are grouped by decade. The prevalence of CH in controls without SCD who were older than 70 years of age is shown in supplemental Figure 4B. Genes carrying the CH mutation in patients with SCD are shown for each age group. In the groups aged 20 to 29, 30 to 39, 40 to 49, 50 to 59, and 60 to 69 years, the percentage of patients with SCD and CH is 0.3%, 1.5%, 3.5%, 5.8%, and 9.1%, respectively, whereas the percentage of controls with CH is 0%, 0.1%, 1.6%, 2.8%, and 5.1%, respectively. N, number; NA, not applicable.
Clinical outcomes were available for 13 patients with CH, with a median follow-up of 6.1 years (range, 0.6-15.2) after blood sampling. The 3 older SCD patients died during follow-up: 2 from solid malignancies (prostate and lung cancer) and the other of unknown cause. Four patients had cardiovascular disease (dilated cardiomyopathy [n = 3] and atherosclerotic disease [n = 1]). No patient developed a hematologic malignancy. Otherwise, these 13 patients presented with other SCD-related complications, including 8 patients with grade 3 or 4 retinopathy and 6 patients with chronic kidney disease. Given our sample size, we cannot conclude whether the presence of CH contributed to these complications. We did not find any significant association between the presence of CH and hematologic parameters (supplemental Table 3).
We report the description of CH mutations in patients with SCD using WES data. As in individuals without SCD,3,4 the frequency of CH increased with age, and DNMT3A was the most frequently mutated gene. We found that the prevalence curve was shifted toward the left in patients with SCD, suggesting an earlier occurrence in this population. However, we point out that this conclusion should not be considered definitive because our study has limitations: the sample size is small, and the WES data sets suffer from heterogeneity in sequencing depth and coverage, although several sensitivity analyses suggest that these differences alone cannot account for our results. In a recent analysis using whole-genome sequencing,21 no difference in CH occurrence was noted between patients and controls. Given these contrasting results and the limited sensitivity of low-depth whole-genome sequencing and WES for detecting CH mutations, there is an urgent need for dedicated analyses using higher-depth targeted sequencing approaches to fully characterize the age-related prevalence and gene spectrum of CH in SCD.
The mechanisms by which SCD may influence the development of CH are unknown. We can postulate that ineffective erythropoiesis and increased erythrocyte turnover may favor the occurrence of a clone carrying a selective advantage,1,22 although many other pathways could be involved (eg, chronic inflammation).2 We identified 2 young patients carrying TP53 clones. These results, when added to those from a recent study,13 raise questions about whether SCD confers a specific advantage to TP53 mutant clones, given the high risk for hematologic malignancy in individuals without SCD.23 No patient developed a hematological malignancy in our study, but the follow-up period for the 2 patients with TP53 clones was short (0 and 2 years). The impact of CH on autologous and allogeneic HCT in patients with SCD warrants further studies. In individuals without SCD, autologous HCT could promote CH and, in the other direction, CH may influence posttransplant mortality and the development of therapy-related myeloid neoplasms.24-26 With regard to gene therapy, it is not known whether CH has an impact on gene transfer, gene editing, and/or outcomes.
Our sample size prevented well-powered analyses of CH genotype-phenotype correlation, as well as analysis of the impact of hydroxyurea on CH occurrence. Notably, dilated cardiomyopathy can have multiple contributing factors in patients with SCD; therefore, it is not possible to draw conclusions on the contribution of CH. Whether CH is a disease-modifying factor warrants further study; however, in our cohort, outcomes of patients with CH were not uniformly unfavorable.
Acknowledgments
The authors thank all SCD participants for their contributions to this project, as well as the participants in the BioMe Biobank for their invaluable contribution to biomedical research. They also thank Gabrielle Boucher for statistical advice and Mélissa Beaudoin, Valérie-Anne Codina-Fauteux, and Sandra Therrien-Laperrière for the amplicon-based validation experiment. Acknowledgments for the CIP data set are in supplemental Data.
This work was supported by the Canadian Institutes of Health Research (PJT #156248), the Doris Duke Charitable Foundation, Bioverativ/Sanofi, and the Canada Research Chair Program (G.L.). T.P. is a recipient of a Charles Bruneau Foundation fellowship award. GEN-MOD sample and data collection were supported by National Institutes of Health/National Heart, Lung and Blood Institute grant HL-68922. The Mount Sinai BioMe Biobank is supported by The Andrea and Charles Bronfman Philanthropies. It is also supported, in part, through the computational resources and staff expertise provided by the Scientific Computing group at the Icahn School of Medicine at Mount Sinai.
Authorship
Contribution: T.P., D.E.B., R.C.L., and G.L. designed the study; T.P., S.L., Y.I., and M.P. performed analyses; T.P. and R.C.L. performed manual variant curation; T.P., A.L.P.H.d’A.d’O., P.B., F.G., P.J., R.J.F.L., and G.L. collected clinical and genetic data; T.P., D.E.B., R.C.L., and G.L. drafted the paper; G.L. supervised the study; and all authors interpreted data, revised the manuscript for critical content, and approved the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Guillaume Lettre, Montreal Heart Institute, 5000 Belanger St, Montreal, QC H1T 1C8, Canada; e-mail: guillaume.lettre@umontreal.ca.
Data sharing requests should be sent to Guillaume Lettre (guillaume.lettre@umontreal.ca).
The online version of this article contains a data supplement.

