

In this issue of Blood, report the first known cases of acute promyelocytic leukemia (APL) caused by integration of the ubiquitous, seemingly nonpathogenic torque teno mini virus (TTMV) into the RARA locus.1
APL accounts for 5% to 10% of pediatric and adult acute myeloid leukemia (AML) and is characterized by specific clinical and biologic features and is managed by a unique therapeutic approach. Indeed, compared with other AML subtypes, it seems APL diagnosis, biology, and therapy are well defined with little left to be worked out. The diagnosis and management of APL often proceeds down a relatively predictable path beginning with the patient presenting with a variably high white blood cell count with promyelocytic blasts, typically with characteristic intense azurophilic granules and obvious Auer rods. Diagnosis is facilitated by the fact that APL is, by in large, a genetically uniform disease characterized by the pathognomonic PML/RARA (promyelocytic leukemia gene/retinoic acid receptor α) fusion gene resulting from the t(15;17)(q22;q12) translocation.2 Importantly, APL is exquisitely sensitive to all-trans retinoic acid (ATRA) and arsenic trioxide (ATO), making it highly curable in most patients without need for intensified chemotherapy.3,4
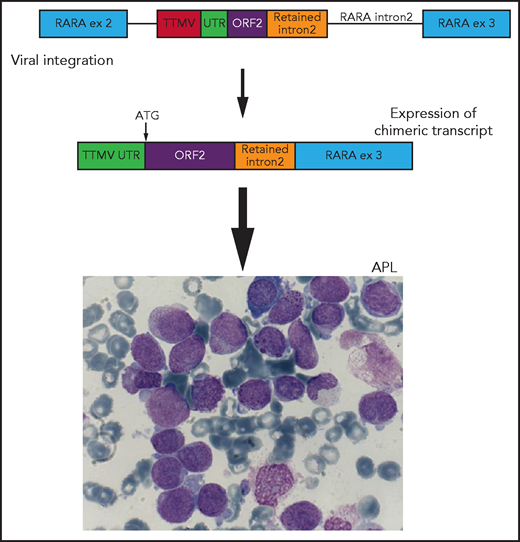
Schematic representation of viral integration of a portion of the TTMV gene into intron 2 of the RARA gene. The integration includes TTMV open reading frame 2 (ORF2) and an upstream untranslated region (UTR) adjacent to a short sequence of retained RARA intron 2, in-frame with exon (ex) 3. This integration results in the transcription of a chimeric fusion messenger RNA with a start codon (ATG) at the initiation of TTMV ORF2. This chimeric fusion transcript results in an APL-like gene expression signature and ultimately the development of acute promyelocytic leukemia (APL). Adapted from Figure 1 in the article by Astolfi et al that begins on page 1773.
Schematic representation of viral integration of a portion of the TTMV gene into intron 2 of the RARA gene. The integration includes TTMV open reading frame 2 (ORF2) and an upstream untranslated region (UTR) adjacent to a short sequence of retained RARA intron 2, in-frame with exon (ex) 3. This integration results in the transcription of a chimeric fusion messenger RNA with a start codon (ATG) at the initiation of TTMV ORF2. This chimeric fusion transcript results in an APL-like gene expression signature and ultimately the development of acute promyelocytic leukemia (APL). Adapted from Figure 1 in the article by Astolfi et al that begins on page 1773.
Of course, APL still poses challenges, including significant disseminated intravascular coagulopathy at presentation that can lead to the patient’s demise from a catastrophic hemorrhage. Because of this risk, physicians suspecting APL must initiate ATRA therapy almost immediately, long before genetic confirmation of the disease. Additionally, patients can suffer life-threatening differentiation syndrome, particularly after ATRA initiation. Beyond these clinical challenges in the early disease course, diagnosis itself can prove surprisingly elusive in some cases that by all clinical and morphologic features fit the definition of APL. Occasionally, the PML/RARA fusion results from a cryptic translocation, missed by all standard cytogenetic assays. Rarely, RARA fusions involving other genes can drive the disease and, in fewer cases still, fusions involving other members of the retinoic acid receptor family have been identified as drivers of APL.5 Astolfi et al report an even more unexpected genetic cause of APL.
The authors report the case of a 6-year-old girl with clinical and morphologic features of APL without evidence of t(15;17) or the PML/RARA fusion. To investigate the genetic cause of her disease after its unfortunate relapse, the authors conducted whole-transcriptome sequencing on a leukemic sample. This analysis identified an in-frame viral insertion of a portion of the TTMV into intron 2 of the RARA gene, within the region of RARA involved in the PML/RARA fusion. This integration resulted in transcription of a chimeric TTMV/RARA fusion gene (see figure). Based on this finding, the patient was treated with ATRA/ATO, which gradually decreased the fusion transcript and ultimately resulted in a complete remission. Going further, the authors interrogated retrospective whole-transcriptome sequencing data, identifying a similar integration of a portion of the TTMV gene into RARA intron 2 in a sample from a 3-year-old with normal karyotype AML. These chimeric fusions were also associated with an APL gene expression signature, and when expressed in a human cell line conferred sensitivity to ATRA, supporting a pathogenic role in the genesis of APL.1
Although viral infections are known drivers of 12% to 16% of cancers, this is the first description of a viral cause of APL.6 Additionally, this is the first direct evidence of TTMV, a nonpathogenic member of the Anelloviridae family of nonenveloped, circular single-stranded DNA viruses, as a cause of cancer. The identification of viral integration causing APL leads one to wonder if viral-driven APL outbreaks are possible, as there are reports of clusters of APL in the literature.7 However, the cases reported in these clusters were characterized by presence of the PML/RARA fusion, thus genetically distinct from the TTMV/RARA fusion cases reported by Astolfi et al. Furthermore, TTMV is an omnipresent virus, likely acquired by all humans during infancy and persisting throughout life8; thus, it is unlikely to be a prominent cause of periodic APL outbreaks. There is evidence suggesting TTMV is held in check by the intact immune system, raising the question of whether increased viral load in an immunocompromised host could facilitate the integration of a portion of the virus into the RARA gene.8 In the 2 cases presented by Astolfi et al, neither child had an identified preexisting immune deficit before their leukemia diagnosis, though this cannot be ruled out.
Last, this fascinating report raises the question of whether TTMV viral integration is a recurrent cause of APL. To fully define the frequency of TTMV/RARA fusions, sequencing of many additional APL samples lacking PML/RARA fusions will be necessary. Given that sensitivity to ATRA and ATO is likely dependent on the presence of the PML/RARA fusion,9 precisely defining the genetic basis of each APL case is of clinical value. Therefore, if future work establishes TTMV/RARA fusion as a recurrent cause of APL, development of clinical assays for its detection could be warranted.
Conflict-of-interest disclosure: The author declares no competing financial interests.

