Key Points
CEBPA-bZip domain mutations are associated with favorable clinical outcomes regardless of monoallelic or biallelic status.
Patients with CEBPA-bZip single- and double-mutant AML have similar clinical and biologic features distinct from those of patients with CEBPA-WT AML.

Abstract
Biallelic CEBPA mutations are associated with favorable outcomes in acute myeloid leukemia (AML). We evaluated the clinical and biologic implications of CEBPA-basic leucine zipper (CEBPA-bZip) mutations in children and young adults with newly diagnosed AML. CEBPA-bZip mutation status was determined in 2958 patients with AML enrolled on Children’s Oncology Group trials (NCT00003790, NCT0007174, NCT00372593, NCT01379181). Next-generation sequencing (NGS) was performed in 1863 patients (107 with CEBPA mutations) to characterize the co-occurring mutations. CEBPA mutational status was correlated with disease characteristics and clinical outcomes. CEBPA-bZip mutations were identified in 160 (5.4%) of 2958 patients, with 132 (82.5%) harboring a second CEBPA mutation (CEBPA-double-mutated [CEBPA-dm]) and 28 (17.5%) had a single CEBPA-bZip only mutation. The clinical and laboratory features of the 2 CEBPA cohorts were very similar. Patients with CEBPA-dm and CEBPA-bZip experienced identical event-free survival (EFS) of 64% and similar overall survival (OS) of 81% and 89%, respectively (P = .259); this compared favorably to EFS of 46% and OS of 61% in patients with CEBPA-wild-type (CEBPA-WT) (both P < .001). Transcriptome analysis demonstrated similar expression profiles for patients with CEBPA-bZip and CEBPA-dm. Comprehensive NGS of patients with CEBPA mutations identified co-occurring CSF3R mutations in 13.1% of patients and GATA2 mutations in 21.5% of patients. Patients with dual CEBPA and CSF3R mutations had an EFS of 17% vs 63% for patients with CEBPA-mutant or CSF3R-WT (P < .001) with a corresponding relapse rate (RR) of 83% vs 22%, respectively (P < .001); GATA2 co-occurrence did not have an impact on outcome. CEBPA-bZip domain mutations are associated with favorable clinical outcomes, regardless of monoallelic or biallelic status. Co-occurring CSF3R and CEBPA mutations are associated with a high RR that nullifies the favorable prognostic impact of CEBPA mutations.
Introduction
CCAAT/enhancer binding protein α (C/EBPα) is a key myeloid transcription factor encoded by the CEBPA gene, and it has a critical role in mediating granulocyte differentiation and cell growth.1-5 Mutations in CEBPA are common drivers for acute myeloid leukemia (AML), occurring in 4% to 11% of patients and enriched in those with normal karyotype.6-11 There are 2 main mutational patterns, and they frequently occur together, typically on separate alleles. One mutational cluster occurs at the N terminus and involves 2 transcription activation domains (TADs; CEBPA-TADs), in which frameshift mutations lead to a truncated translational product because of the use of the alternate start codon.6,12 Mutations in the N-terminal TAD region can occur as germline events, resulting in a predisposition to developing AML.13-15 The other mutational cluster occurs at the C terminus, involves the basic leucine zipper (bZip) region, and often includes in-frame indels that disrupt the dimerization and DNA binding function of the protein.
Cooperation of the bZip and TAD mutations is considered a potent leukemogenic event in AML, and the biallelic acquisition of these mutations has been associated with a favorable prognosis. The current World Health Organization (WHO) AML classification considers a biallelic CEBPA mutation (CEBPA-double-mutated [CEBPA-dm]) as a distinct entity.16 We have previously shown that single TAD mutations are exceptionally rare events in pediatric AML; in an analysis of a limited subset, patients with an isolated bZip mutation (CEBPA-bZip) were shown to have a favorable prognosis comparable to that of patients with CEBPA-dm.7 This study validates our previous observation on the favorable impact of CEBPA-bZip across 4 different clinical trials and provides clinical outcome data for cooperating mutations in patients with CEBPA-mutant AML.
Methods
Patients and treatment
Children and young adults (age 0-29.9 years) with de novo AML enrolled in 4 consecutive Children’s Cancer Group (CCG)/Children’s Oncology Group (COG) trials—CCG2961 (NCT00003790), AAML03P1 (NCT0007174), AAML0531 (NCT00372593), and AAML1031 (NCT01379181)—were eligible for this study. A total of 2958 patients with available biologic and clinical data were included in our study (CCG2961, n = 552; AAML03P1, n = 266; AAML0531, n = 917; AAML1031, n = 1233). Details of these trials have been previously described, and treatment of patients with CEBPA mutations is included in supplemental Methods (available on the Blood Web site).17-20 The presence of a biallelic or any CEBPA mutation was not used for risk stratification or in assigning treatment allocation in CCG2961, AAML03P1, or AAML0531. Risk stratification in AAML1031 stratified patients with any CEBPA-bZip mutation as low risk. The protocols were approved by the institutional review boards of all participating centers and conducted in accordance with the Declaration of Helsinki.
Assessment of mutational status
For patients enrolled on CCG2961 and AAML03P1, polymerase chain reaction amplification of the entire coding region of CEBPA was performed as previously described.7,11 Given the paucity of CEBPA-TAD mutations observed in these trials, subsequent trials analyzed the bZip domain of CEBPA by fragment length analysis, and patients with detectable mutations underwent Sanger sequencing for identification and verification of the specific mutation. In all patients in whom bZip mutations were detected, the N-terminal region was sequenced for identification of TAD mutations. A subset of patients (n = 1863), including n = 110 with CEBPA mutations, underwent comprehensive next-generation sequencing (NGS), which was performed by using target alignment of transcriptome (n = 1056), targeted gene capture (n = 786), or whole-genome sequencing (n = 206), as described previously.21
Transcriptome analysis
Transcriptome data were available from 60 patients with CEBPA-dm, 13 with CEBPA-bZip, and 1436 with CEBPA-wild-type (WT) enrolled on AAML0531 and AAML1031. The data were generated by the British Columbia Genome Sciences Center (BCGSC; Vancouver, BC, Canada). Unsupervised clustering and differential gene analyses were performed on CEBPA-WT and mutated samples. Total RNA samples were ribodepleted and prepared for sequencing using a strand-specific messenger RNA (mRNA) library construction protocol. Indexed libraries were then pooled and sequenced on an Illumina Hiseq sequencing system that produced 75-bp paired-end sequence reads. Sequencing data were aligned to human genome assembly GRCh37. Gene-level counts were quantified using BCGSG’s in-house pipeline and annotated using Ensembl v69 annotations. All transcriptome analysis was performed in the R statistical environment (v3.6.0/v3.6.1).
Unsupervised clustering of the patients with CEBPA mutations and CEBPA-WT was performed via uniform manifold approximation and projection (UMAP) and hierarchal clustering. For UMAP clustering, gene counts were size-factor scaled by the geometric mean of the total read counts per sample, followed by term frequency-inverse document frequency transformation. Input genes for clustering (6816 genes) were selected by using the mean vs dispersion parametric model trend (SeqGlue v0.1) followed by jackstraw principal component analysis (jackstraw v1.3) to identify genes significantly associated with the first 20 principal components. UMAP was carried out with the UWOT v0.1.5 (https://arxiv.org/abs/1802.03426v2), and Leiden clustering of the UMAP reduced dimension data was performed with SeqGlue v0.1.
Log2-transformed and scaled counts of the previously selected 6816 genes were used as input for unsupervised hierarchical clustering of the CEBPA-mutated patients via Ward’s hierarchical clustering method (ComplexHeatmap v2.0.0). Differential expression analysis was performed using edgeR (v3.26.5) and limma (v3.40.5). Differentially expressed genes with a log-fold change of greater than +1 or –1 were retained, and a false discovery rate–adjusted threshold of P < .05 was used to determine significance. Further identification of enriched pathways was performed via active subnetwork analysis of the log-fold changes and P values obtained from limma (pathfindR v1.4.2).
Statistical analyses
The Kaplan-Meier method was used to estimate overall survival (OS), event-free survival (EFS), disease-free survival (DFS), and relapse risk (RR).22 OS was defined as the time from study entry to death as a result of any cause or date of last follow-up in surviving patients; EFS as the time from study entry until induction failure, relapse, or death; DFS as the time from end of course 1 for patients in complete remission (CR) until relapse or death or date of last follow-up for those without an event; and RR as the time from end of induction 1 for patients in CR to relapse in which deaths without a relapse were considered competing events.23 CR was defined as a bone marrow aspirate containing <5% blasts by morphology and without evidence of extramedullary disease. Patients shown to be in a remission without evidence of measurable residual disease (MRD) were considered to be MRD-negative and were defined as having a bone marrow aspirate containing <0.1% blasts detected by flow cytometry. The Kruskal-Wallis test was used for comparison of continuous variables, and the χ2 test was used to test the significance of observed differences in proportions. Fisher’s exact test was used when the sample size was small.
Results
CEBPA mutation status and correlation with disease characteristics
Mutations in the bZip domain were identified in 160 (5.4%) of 2958 patients. Among patients with bZip mutations, 132 (82.5%) harbored a cooperating TAD domain mutation on the other allele, and the remaining 28 patients (17.5%) lacked a second CEBPA mutation (supplemental Figure 1). Sequencing of the full coding region of CEBPA was performed in 2607 patients (88% of the cohort); a total of 3 patients were identified by NGS and 1 by conventional sequencing as harboring CEBPA-TAD mutations only, for a total prevalence of 0.15%. Given the overall paucity of these mutations in our cohort, these patients were excluded from all further analyses. Analysis according to demographics demonstrated similar distribution across studies and among patients with CEBPA-dm, CEBPA-bZip, and CEBPA-WT (Table 1). Patients with CEBPA mutations were older (median age: CEBPA-WT, 9.5 years; CEBPA-bZip, 12.3 years; CEBPA-dm, 14 years; P < .001) and had higher diagnostic white blood cell counts (WBC) (median WBC: CEBPA-WT, 21.8 × 103/μL; CEBPA-bZip, 45.3 × 103/μL; CEBPA-dm, 39.4 × 103/μL; P < .001; Table 1). Analysis of the prevalence of CEBPA mutations among age-defined cohorts (young children, age 0-9 years; older children, age 10-15 years; adolescents and young adults, age 16-29 years) demonstrated that CEBPA-bZip mutations were associated with the younger age cohort, accounting for 32% of CEBPA mutations in this group (P = .016; Table 1).
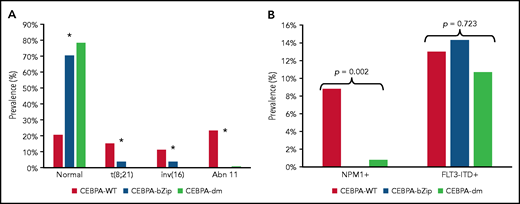
Complete cytogenetic data were available for 2543 patient samples (86%) (Table 1). Patients with a CEBPA mutation were more likely to have a normal karyotype compared with CEBPA-WT (CEBPA-WT, 20.5%; CEBPA-bZip, 70.4%; CEBPA-dm, 78.3%; P < .001; Figure 1A). There was a paucity of cytogenetic alterations such as t(8;21), inv(16), and chromosome 11 abnormalities, specifically KMT2A rearrangements, among CEBPA-mutant compared with patients with CEBPA-WT (Figure 1A). There were no differences in the prevalence of cytogenetic alterations between patients with CEBPA-bZip and those with CEBPA-dm (Figure 1). NPM1 and FLT3-internal tandem duplication (FLT3-ITD) status were available in 97% and 99.5% of patients, respectively. NPM1 mutations were nearly absent in patients with CEBPA mutations (CEBPA-WT, 8.8%; CEBPA-bZip, 0%; CEBPA-dm, 0.8%; P = .002), and there was a similar prevalence of FLT3-ITD (allelic ratio [AR] >0.1) across the groups (CEBPA-WT, 13%; CEBPA-bZip, 14.3%; CEBPA-dm, 10.7%; P = .723; Figure 1B).
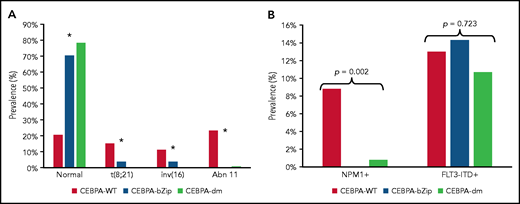
Cytogenetic and molecular characteristics according to CEBPA mutational status. (A) Cytogenetic profile according CEBPA-WT compared with CEBPA-bZip or CEBPA-dm status. *P < .001 for CEBPA-WT compared with CEBPA-bZip and CEBPA-dm. (B) Molecular profile according to CEBPA-WT compared with CEBPA-bZip or CEBPA-dm status. Abn 11, chromosome 11 abnormalities.
Cytogenetic and molecular characteristics according to CEBPA mutational status. (A) Cytogenetic profile according CEBPA-WT compared with CEBPA-bZip or CEBPA-dm status. *P < .001 for CEBPA-WT compared with CEBPA-bZip and CEBPA-dm. (B) Molecular profile according to CEBPA-WT compared with CEBPA-bZip or CEBPA-dm status. Abn 11, chromosome 11 abnormalities.
NGS data were available for 1863 patients, including 107 patients (67%) with CEBPA bZip (single or double) mutations, and this cohort was used for more comprehensive characterization of co-occurring mutations. Cooperating molecular mutations were detected in 70.1% (n = 61) of CEBPA-dm patients and 60% (n = 12) of CEBPA-bZip patients (Figure 2). Mutations in GATA2 and CSF3R genes were highly enriched and mutually exclusive among patients with CEBPA mutations with a prevalence of 21.5% and 13.1% compared with 1.0% and 1.1% in patients with CEBPA-WT, respectively (P < .0001 for both; Figure 2B). Prevalence of NRAS and FLT3-ITD mutations were similar between patients who were CEBPA-mutant and CEBPA-WT (29.0% vs 30.4% and 11.2% vs 12.8%, respectively; P > .05 for both; Figure 2B). Among patients with CEBPA-dm and CEBPA-bZip, there were no significant differences in prevalence in the most common co-occurring mutations (GATA2, CSF3R, NRAS, FLT3-ITD, WT1; P > .05 for all).
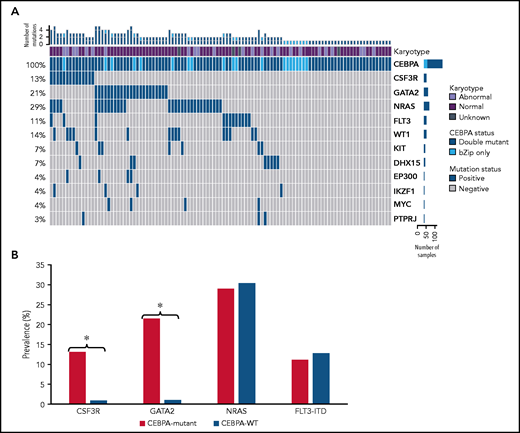
Molecular profiles of patients with CEBPA mutations as determine by NGS. (A) Cyto-molecular status and co-occurring mutational profile of patients with CEBPA double and CEBPA-bZip mutations. (B) Comparison of common co-occurring mutations in patients with CEBPA mutations compared with patients with CEBPA-WT. *P < .0001.
Molecular profiles of patients with CEBPA mutations as determine by NGS. (A) Cyto-molecular status and co-occurring mutational profile of patients with CEBPA double and CEBPA-bZip mutations. (B) Comparison of common co-occurring mutations in patients with CEBPA mutations compared with patients with CEBPA-WT. *P < .0001.
CEBPA status and correlation with clinical outcome
Patients with CEBPA mutations had a favorable initial response to therapy with a CR rate of 87.7% compared with 76.9% in patients with CEBPA-WT (P = .002). Those with CEBPA-dm had a CR rate of 89.8% compared with 78.6% in CEBPA-bZip patients (P = .116; Table 1). End-of-induction MRD status was available in 2086 (86.8%) patients treated on AAML03P1, AAML0531, and AAML1031 (70.6% of the cohort overall). The MRD-negative CR rate was 83.4% for patients with CEBPA mutations compared with 70.5% for patients with CEBPA-WT (P = .002). Analysis according CEBPA mutation type demonstrated similar MRD-negative CR rates among patients with CEBPA-dm and CEBPA-bZip of 82.5% and 87.5%, respectively (P = .761; Table 1).
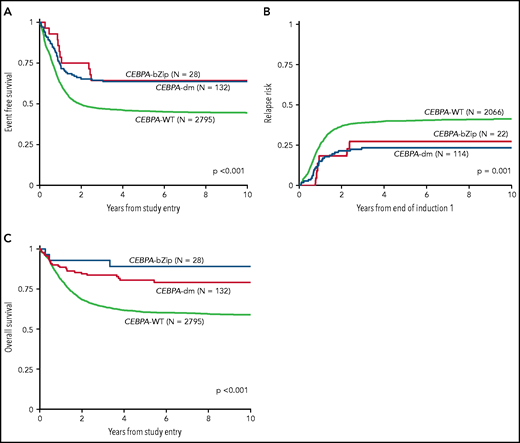
Analysis of the overall cohort according to the type of CEBPA mutation demonstrated that those with CEBPA-dm and CEBPA-bZip experienced identical 5-year EFS of 64% ± 9% and 64% ± 18%, respectively (P = .777; Figure 3A), which were both superior compared with that of patients with CEBPA-WT (5-year EFS, 46% ± 2%; P < .001; Figure 3A). Corresponding 5-year RRs were similar among patients with CEBPA-dm (23% ± 8%) and CEBPA-bZip (27% ± 20%; P = .765) and were lower than those in patients with CEBPA-WT (40% ± 2%; P = .001; Figure 3B). Corresponding 5-year OS was also similar for CEBPA-dm (81% ± 7%) and CEBPA-bZip (89% ± 12%; P = .259) patients and compared favorably to that for patients with CEBPA-WT (61% ± 2%; P < .001; Figure 3C). Analysis across the age cohorts demonstrated no differences in 5-year OS, EFS, or RR between patients with CEBPA-bZip and CEBPA-dm (supplemental Table 2). Given that there were changes in treatment over time, we analyzed the outcome of patients with CEBPA-dm and CEBPA-bZip treated on the successive studies and found that they experienced similar outcomes that compared favorably to those of CEBPA-WT patients in all of the trials evaluated (supplemental Figure 2). Although the use of allogeneic hematopoietic stem cell transplantation (HSCT) vs chemotherapy for treating patients, including those with CEBPA mutations, has evolved over the course of the studies we evaluated, only a minority of patients with CEBPA mutations were allocated to receive allogeneic HSCT (11.3%), and this was similar over the 4 studies evaluated (range, 7% to 15.7%; supplemental Table 1).
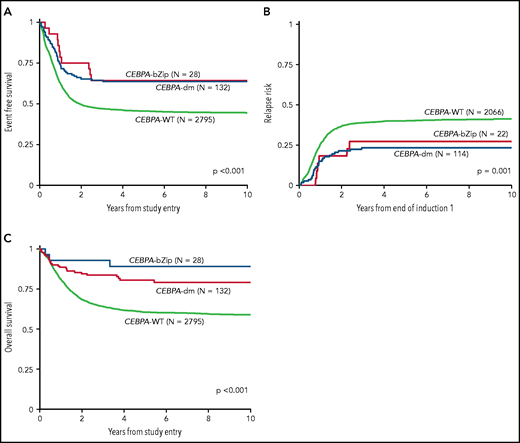
Correlation of CEBPA mutational status with clinical outcome. (A) EFS among the cohort according to CEBPA-WT vs CEBPA-dm vs CEBPA-bZip status; (B) RR according to CEBPA-WT vs CEBPA-dm vs CEBPA-bZip status; (C) OS for the cohort according CEBPA-WT vs CEBPA-dm vs CEBPA-bZip status.
Correlation of CEBPA mutational status with clinical outcome. (A) EFS among the cohort according to CEBPA-WT vs CEBPA-dm vs CEBPA-bZip status; (B) RR according to CEBPA-WT vs CEBPA-dm vs CEBPA-bZip status; (C) OS for the cohort according CEBPA-WT vs CEBPA-dm vs CEBPA-bZip status.
Patients with CEBPA mutations experienced high CR rates; therefore, we subsequently evaluated the impact of achieving MRD on overall outcomes. Among patients with CEBPA mutations with evaluable MRD data (n = 121), the RR according to MRD status was 30% ± 10% for MRD-negative (n = 101) compared with 23% ± 25% for MRD-positive patients (n = 20; P = .595). Corresponding DFS for MRD-negative patients was 67% ± 10% (OS, 87% ± 7%) compared with 62% ± 27% (OS, 79% ± 19%) for patients who were MRD positive (DFS: P = .636; OS: P = .105).
Transcriptome profiling of CEBPA mutations
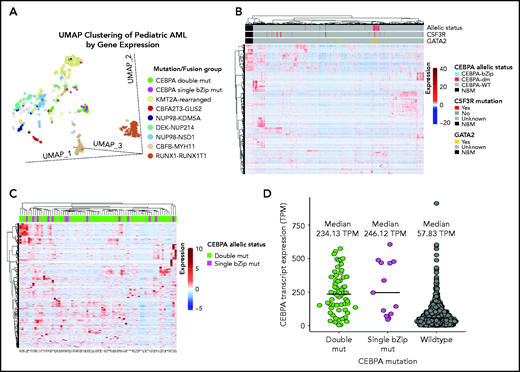
Given the lack of any clear biologic or clinical differences, we inquired whether patients with CEBPA-dm (n = 60) or CEBPA-bZip (n = 13) have distinct gene and mRNA expression profiles by evaluating available transcriptome data and comparing their profiles to those of patients with CEBPA-WT (n = 1436). Using UMAP to compare gene expression profiles of RNA-sequencing data demonstrated that a majority of patients with CEBPA mutations (54 [74%] of 73) clustered together with shared cluster membership and were distinct from patients with other molecular alterations (P < .0001; Figure 4A). In the CEBPA-mutant cluster, no significant differences were detected in cluster membership between CEBPA-bZip (8 of 13) and CEBPA-dm (46 of 60) samples (P = .43). Unsupervised hierarchical clustering of the transcriptome using the same set of genes as UMAP clustering demonstrated distinct expression profiles and differential clustering of patients with CEBPA mutations compared with patients with CEBPA-WT (Figure 4B). Within the group of patients with CEBPA mutations, those with CEBPA-bZip or CEBPA-dm demonstrated a similar mRNA expression profile (Figure 4C). Evaluation of CEBPA transcript expression, measured in transcripts per million (TPM), found that expression was similar among patients with CEBPA-dm (median, 234.13; range, 3.58-574.22) and those with CEBPA-bZip (median, 246.12; range, 45.61-605.56; P = .78). In contrast, patients with CEBPA-WT had significantly lower transcript expression (median, 58.98; range, 0.13-911.28) compared with patients with CEBPA mutations (P < .0001; Figure 4D).
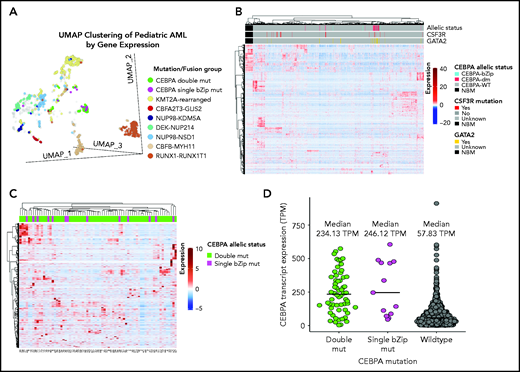
Transcriptome profiling according to CEBPA mutational status. (A) Gene expression clustering according to mutational status with CEBPA-mutant samples compared with other molecular alterations that have prognostic implications in pediatric AML. (B-C) Unsupervised hierarchical clustering of patients according to CEBPA mutational status (B) and with CEBPA-bZip and double mutations (C). (D) CEBPA mutational status and association with CEBPA transcript expression. mut, mutation; TPM, transcripts per million.
Transcriptome profiling according to CEBPA mutational status. (A) Gene expression clustering according to mutational status with CEBPA-mutant samples compared with other molecular alterations that have prognostic implications in pediatric AML. (B-C) Unsupervised hierarchical clustering of patients according to CEBPA mutational status (B) and with CEBPA-bZip and double mutations (C). (D) CEBPA mutational status and association with CEBPA transcript expression. mut, mutation; TPM, transcripts per million.
Clinical significance of cooperating mutations
We inquired whether co-occurrence of the highly enriched molecular variants of CSF3R and GATA2, which were mutually exclusive in the setting of CEBPA-mutant AML, had an impact on the clinical significance of CEBPA mutations. Analysis according to CSF3R status, excluding patients with GATA2 mutations, demonstrated that co-occurrence of a CSF3R mutation significantly modulated the favorable EFS conferred by CEBPA. Patients with dual CEBPA+/CSF3R+ mutations experienced an EFS of 23% ± 23% compared with 62% ± 12% in patients with CEBPA+/CSF3R-WT (P = .002; Figure 5A). This disparate outcome in the patients with CEBPA+/CSF3R+ was driven by a higher RR of 77% ± 26% compared with 23% ± 11% in patients with CEBPA+/CSF3R-WT (P < .001; Figure 5B). Despite the higher RR, patients with CEBPA+/CSF3R+ achieved an OS comparable to that of patients with CEBPA+/CSF3R-WT (76% ± 25% vs 84% ± 9%, respectively; P = .644; Figure 5C), demonstrating that salvage therapy could be used successfully after patients relapsed. In contrast, analysis according to GATA2 status, excluding patients with CSF3R mutations, demonstrated that co-occurrence of GATA2 had no impact on clinical outcomes; patients with CEBPA+/GATA2+ and those with CEBPA+/GATA2-WT demonstrated similar 5-year EFS (70% ± 19% vs 62% ± 12%; P = .543), RR (15% ± 16% vs 23% ± 11%; P = .431), and OS (86% ± 16% vs 84% ± 9%; P = .988; Figure 5).
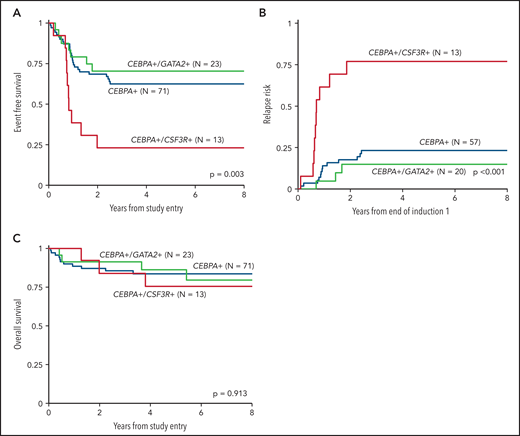
Outcomes of CEBPA-mutant patients according to co-occurring CSF3R and GATA2 mutational status. (A) EFS of dual CEBPA+/CSF3R+ mutant patients compared to those with dual CEBPA+/GATA2+ and those with a CEBPA+ mutation and neither CSF3R nor GATA2; (B) RR of dual CEBPA+/CSF3R+ mutant patients compared to dual CEBPA+/GATA2+ and patients with a CEBPA+ mutation and neither CSF3R nor GATA2; (C) OS of dual CEBPA+/CSF3R+ mutant patients compared to dual CEBPA+/GATA2+ and patients with a CEBPA+ mutation and neither CSF3R nor GATA2.
Outcomes of CEBPA-mutant patients according to co-occurring CSF3R and GATA2 mutational status. (A) EFS of dual CEBPA+/CSF3R+ mutant patients compared to those with dual CEBPA+/GATA2+ and those with a CEBPA+ mutation and neither CSF3R nor GATA2; (B) RR of dual CEBPA+/CSF3R+ mutant patients compared to dual CEBPA+/GATA2+ and patients with a CEBPA+ mutation and neither CSF3R nor GATA2; (C) OS of dual CEBPA+/CSF3R+ mutant patients compared to dual CEBPA+/GATA2+ and patients with a CEBPA+ mutation and neither CSF3R nor GATA2.
Co-occurring FLT3-ITD mutations (AR >0.1) were detected in 17 (11%) of 149 patients with CEBPA mutations, with an AR range of 0.14 to 0.97. Outcome data were available in 15 patients with dual CEBPA+/FLT3-ITD+, and analysis demonstrated that they had similar EFS and OS compared with patients with CEBPA mutations who lacked an ITD mutation (supplemental Figure 3).
Discussion
In this large cohort of 2948 children and young adults with newly diagnosed AML, we demonstrated that patients with CEBPA mutations that had single bZip domain mutations experienced outcomes nearly identical to those of patients with biallelic CEBPA mutations. Our findings align with those of Georgi et al24 who reported on a cohort of 4578 adult patients with AML and showed analogous outcomes for patients with CEBPA-dm and CEBPA-bZip. Our study provides more definitive support that the presence of a CEBPA-bZip mutation is associated with favorable outcome, regardless of monoallelic or biallelic status.
Given the lower prevalence of CEBPA-bZip only mutations, previous studies have not been powered to evaluate the prognostic significance of this subset separately. Dufour et al25 found that patients with CEBPA-dm had a superior median OS compared with that of patients with CEBPA-WT, whereas patients with monoallelic CEBPA mutations had outcomes comparable to those of patients with CEBPA-WT. However, the majority of the patients in the monoallelic CEBPA cohort harbored a CEBPA-TAD mutation, and they did not analyze CEBPA-TAD and CEBPA-bZip mutation cohorts separately. Analyses that combined both cohorts likely obscured any differences between CEBPA-TAD and CEBPA-bZip and may explain the contrast seen in our results.8,9,25-27 Analysis of a uniform monoallelic CEBPA-TAD mutation population by Georgi et al24 demonstrated no prognostic impact.
C/EBPα exists in 2 translational isoforms (p42 and p30) that dimerize via various combinations into a transcription factor essential for normal monopoiesis and granulopoiesis.28CEBPA-TAD frameshift mutations induce an early termination codon that leads to the generation of a truncated protein (p30 isoform) which, although it is missing the TAD that is key to transcriptional regulatory activity, retains functional DNA binding. In addition to quantitative deficit of the p42 isoform, the excess p30 isoform acts as a dominant negative on any remaining p42 isoform.6,12 In contrast, CEBPA-bZip in-frame insertions or deletions occur at the junction between the basic region and the leucine zipper, which leads to a qualitative deficit by disrupting the DNA binding and/or dimerization of both isoforms.6,12 Although germline CEBPA-TAD mutations result in a predisposition to AML,13,14 acquisition of somatic CEBPA-bZip mutations have been detected in nearly all patients who progress to AML.15,29 This implies that CEBPA-TAD and the resultant truncated CEBPA isoform are dependent on additional CEBPA mutations as well as mutations in other oncogenic genes for leukemogenic transformation. The biallelic presence of TAD and bZip mutations cooperate to create a highly penetrant malignant phenotype resulting in loss of DNA binding in the full-length isoform; thus, the truncated isoform is the only functional product. Although this cooperativity is highly leukemogenic, our findings show that the presence of a monoallelic bZip mutation and the resultant disruption in DNA binding is sufficient for association with favorable outcome.
Our results also demonstrate that patients with CEBPA-dm and CEBPA-bZip share similar biologic features. The 2 groups have similar cytogenetic and molecular characteristics and harbor significant differences compared with patients with CEBPA-WT. Similarly, Georgi et al24 found an overlapping molecular profile between CEBPA-dm and CEBPA-bZip patients distinct from that of patients with CEBPA-TAD. Specifically, patients with CEBPA-dm and CEBPA-bZip in their study had a high prevalence of GATA2 mutations. An association between CEBPA-TAD mutations and genes involved in epigenetic regulation has been described in adult groups, including those in the study by Georgi et al, but both of these types of events are exceedingly rare in pediatrics.24,30,31 We demonstrate that patients with CEBPA-bZip and CEBPA-dm shared similar gene expression and transcriptome profiles that were distinct from those of patients with CEBPA-WT. These findings suggest that the presence of the bZip mutation and the resultant deficient DNA binding and dimerization may have a significant impact on transcription, including critical myeloid lineage–affiliated genes. Although previous studies suggested that patients with CEBPA-dm had a distinct transcriptome profile compared with patients who had single mutations, those analyses included single TAD as well as bZip variants in the single-mutation cohort.8,27 Our data clearly demonstrate that even though patients with CEBPA mutations have a unique transcriptome profile, there are no significant distinguishing features between patients with CEBPA-dm and CEBPA-bZip only. This further substantiates lack of biologic distinction between CEBPA-dm and CEBPA-bZip AML.
The striking enrichment of CSF3R and GATA2 mutations in patients with CEBPA mutations is intriguing and aligns with previously reported findings. CSF3R mutations are rare, but when they are compared with mutations in adult AML, they appear slightly more prevalent in childhood AML and almost exclusively occur in the setting of CEBPA mutations or RUNX1-RUNX1T1 fusions.21,32,33 Braun et al34 demonstrated functional significance in the order of acquisition with the initial acquisition of a CEBPA mutation required for a subsequent CSF3R mutation to have proliferative impact. In this study, we demonstrated that the presence of a CSF3R mutation in patients with CEBPA mutations is associated with a remarkably high RR and poor EFS. However, despite such high RR, patients with dual CEPBA+/CSF3R+ experience OS similar to that of patients with CSF3R-WT. Because allogeneic HSCT is considered standard treatment for relapse in pediatric AML, our data suggest that patients with CEPBA+/CSF3R+ are responsive to intensified therapy. Given the poor response to first-line chemotherapy, intensifying treatment early in these patients and/or consolidating a first remission with HSCT warrants further consideration. The overlap of CEBPA and GATA2 mutations is well recognized in adult AML,35,36 and our findings in pediatric AML further support mechanisms of cooperativity. Furthermore, the neutral prognostic effects are consistent with previous studies in adult AML.36-38
The findings we present regarding the favorable outcomes across the studies suggest that bZip mutations, regardless of their biallelic status, retain prognostic impact across the different treatment regimens. Presence of a CEBPA-bZip mutation was used to classify patients as favorable risk in the AAML1031 trial, and they were allocated to receive chemotherapy unless high allelic ratio FLT3-ITD or refractory disease was present, but patients with CEBPA mutations treated on the predecessor trials were allocated to receive allogeneic HSCT if a matched family donor was available. However, we do not think the differences in use of allogeneic HSCT as first-line therapy confounded our findings because a significant majority of patients with CEBPA mutations in our cohort were treated with chemotherapy, and HSCT rates were similar across the different studies. We also report that patients with CEBPA mutations, regardless of MRD status after induction 1, experienced similar and favorable outcomes. It has previously been reported that favorable-risk pediatric patients (NPM1, CEBPA, inv(16)/t(16;16), t(8;21)) who are MRD-positive experience relapse-free survival nearly identical to that of patients who are MRD-negative.39 In addition, favorable-risk patients have been shown to benefit from intensification of chemotherapy, suggesting that the biology of favorable-risk AML may be more permissive to the cytotoxic effects of chemotherapy.19,40 We hypothesize that because patients with CEBPA mutations generally exhibit chemotherapy-sensitive disease (even those in CR but with persistent MRD after induction 1), subsequent intensive chemotherapy with a second intensive induction and multiple high-dose cytarabine consolidation courses can achieve durable remissions.
Our findings are also in line with our previous observations on the paucity of single TAD mutations in pediatric AML and are further supported by work from Georgi et al24 that did not identify single TAD mutations in patients age 30 years or younger in a large cohort and also found that those mutations were not associated with outcome.7,24 Nonpathogenic polymorphisms in the TAD domains have also been identified and are important for differentiation from pathogenic mutations.7,41,42 We did not sequence the full coding region of CEBPA in our entire cohort using NGS and conventional sequencing, but we did screen for TAD mutations in a large majority (88%) of patients and found that those with single TAD mutations were rarely identified (0.15%). Even with our large sample size, a pediatric CEBPA-TAD cohort large enough for comparisons to be adequate could not be generated. In addition, on the basis of our results demonstrating the rarity of single TAD mutations in pediatric AML, it is exceedingly unlikely that occult single TAD mutations would have been misclassified as CEBPA-WT and would have obscured any impact on this group.
CEBPA-dm is incorporated as a distinct entity into the WHO classification of myeloid neoplasms and leukemia; CEBPA-dm is now considered a favorable prognostic feature. Past observations regarding the prognostic significance of monoallelic CEBPA mutations compared with CEBPA-dm have yielded conflicting results. Importantly, our findings demonstrate that patients with a bZip mutation, either monoallelic or biallelic, have favorable outcomes and similar biology; this provides a strong rationale for modifying the current WHO guidelines to broaden the requirement for the presence of CEBPA-dm as a prognostic entity to a CEBPA-bZip mutation.
Acknowledgments
This work was supported by grants from the St Baldrick’s Foundation (St Baldrick’s Scholar [K.T.], St Baldrick’s Consortium Grant [S.M.]), Target Pediatric AML (S.M.), Leukemia and Lymphoma Society (6558-18, S.M. and E.A.K.); by grants from the National Cancer Institute, National Institutes of Health (R01-CA114563-10) (S.M.), the Children's Oncology Group (COG) Chair (U10-CA098543) (S.M.), the National Clinical Trials Network (NCTN) Statistics and Data Center (U10-CA180899) (S.M. and T.A.A.), and the NCTN Operations Center (U10CA180886) (S.M. and E.A.K.); and by grants from the US Department of Health and Human Services (HHSN-261200800001E) (S.M.), the Andrew McDonough B+ Foundation (S.M.), Hyundai Hope on Wheels (S.M.), and Project Stella (S.M.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: K.T., A.J.L., A.L., J.L.S., and R.E.R. prepared the figures; K.T., A.J.L., T.A.A., Y.-C.W., R.B.G., R.E.R., A.L., J.L.S., L.P., L.E.B., and S.M. contributed and analyzed the data; M.R.L., W.G.W., T.M.C., E.A.K., A.S.G., and R.A. provided general scientific guidance; K.T., A.J.L., and S.M. wrote the manuscript; and all authors reviewed and contributed to the manuscript before it was submitted.
Conflict-of-interest disclosure: M.R.L., L.E.B., and L.P. are employees of Hematologics, Inc., and M.R.L. has equity ownership in Hematologics, Inc. The remaining authors declare no competing financial interests.
Correspondence: Katherine Tarlock, Division of Hematology/Oncology, Seattle Children’s Hospital, University of Washington, M/S MB.8.501, PO Box 5371, Seattle, WA 98145-5005; e-mail: katherine.tarlock@seattlechildrens.org.
For original data, please contact the corresponding author.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.